

The Role of the Cardiologist in PAH Diagnosis and Management
Vous trouverez des articles traitant du même sujet dans la catégorie "HTAP"
Problèmes que peuvent renconter les patients atteints de la maladie de Gaucher
Liens utiles à la fin des catégories.
Ghislaine SURREL
maladies-lysosomales-subscribe@yahoogroupes
![]()
Michael A. Mathier, MD, FACC

Slide 1. The Role of the Cardiologist in PAH Diagnosis and Management
I am sure this will be a very interesting and informative session, dealing with what can accurately be described as the most rapidly evolving field in all of cardiovascular medicine, and that is pulmonary arterial hypertension (PAH). It also is an interesting field because it is largely shared by cardiology and pulmonary, and my goal today is to give you my idea of what I think the essential features of the role of the cardiologist is in the diagnosis and management of this disease.

Slide 2. The Role of the Cardiologist in PAH: Summary
Here is the summary of my thoughts on this, and I would like to go through them. Then we are going to spend a couple of minutes on each of these throughout the body of the talk.

Slide 3. The Role of the Cardiologist in PAH
The first is that I think it is important that we all have at least a working knowledge of this disease state, that we maintain a high index of suspicion and, as I will point out a little later, we are going to see an awful lot of patients in our office practices who present with very nonspecific signs and symptoms. And if we are going to make a diagnosis of PAH, it has to be from a foundation of a high index of suspicion. We must have an informed ability to interpret the screening studies that are used in this disease state. We carry an expertise beyond that of many of our colleagues, and we must be able to apply that expertise, both for our own practice, and also to help our colleagues understand the results they are getting from the initial workup for this disease. Importantly, we must be willing to perform and properly interpret right-heart catheterization and hemodynamic assessment in patients undergoing evaluation for this disease. We must be in a position where we can treat this disease to a degree that is commensurate with our interest and our understanding of it. It is not going to be a disease state that everybody embraces in terms of treatment, but everybody needs to understand what the disease state is, and at the very least, be able to channel patients towards proper therapy. Importantly, have a careful eye out for the development of right ventricular (RV) failure and participate in its diagnosis and management, and then, as always, be vigilant for superimposed cardiovascular diseases.
A working knowledge of the disease -- what do we mean by this?

Slide 4. Pulmonary Arterial Hypertension: Definition
A definition of PAH is shown here; this is probably the most commonly employed definition. As you can see, it is a hemodynamic one. It is the presence of a mean pulmonary pressure in excess of 25 mm Hg simultaneous to a wedge pressure that is normal or near normal. Remember that pulmonary hypertension (PH) on its own is defined as an elevation in the pulmonary arterial (PA) pressure, but if you are going to specify PAH, it needs to be at the same time that the left-sided cardiac filling pressure is normal or near normal.
Because we now have a number of very good therapies for this disease state, there has been increasing interest in trying to make the diagnosis earlier, and one method that has been put forward is the measurement of PA pressures during exercise. I wouldn't say that this has become widely accepted or routinely performed, but there has been the suggestion that an exercise mean PA pressure in excess of 35 mm Hg might identify a patient population with early disease.

Slide 5. World Conference on Pulmonary Hypertension Revised Nomenclature and Classification (Venice 2003)
I think it is important that we know that the nomenclature and classification of this disease have been relatively recently updated. Rather than go through the entire list, what I would like to point out is that the first group outlined here, so-called World Health Organization (WHO) Group 1, represents on the surface a heterogeneous group of diseases, but in fact a group of diseases that share common pathophysiology, and importantly, share common response to the specific PH therapies that Dr. McLaughlin is going to talk about. These include idiopathic PAH; what used to be called primary pulmonary hypertension (PPH), the familial form; the forms associated with rheumatologic disorders or collagen vascular diseases; congenital heart disease; portopulmonary hypertension; human immunodeficiency virus (HIV)-related PH; PH associated with drugs or toxins including anorexogens; and a rare disorder, pulmonary veno-occlusive disease (PVOD).
We see tons of the other classifications, but they are distinct from what we are talking about. The bulk of what we are talking about lies in the WHO Group 1 classification.
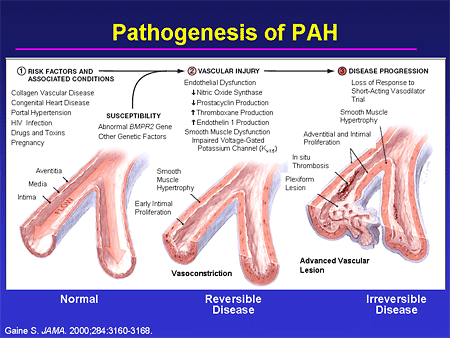
Slide 6. Pathogenesis of PAH
It is also important to recognize, without getting lost in the detail, that we now have a remarkably better understanding of who is at risk for this disease. We are starting to understand more and more about genetic susceptibility, and we have identified a number of signaling pathways that become disordered in this disease state. This has, importantly, led to the development of a host of therapeutic options. What we are hoping now is that our understanding of this entire progression is going to allow us to identify patients earlier in the disease state, and allow us to either delay or perhaps prevent progression to endstage disease, and the awful prognosis that that still carries.

Slide 7. The Role of the Cardiologist in PAH
High index of suspicion.

Slide 8. Assessment of PAH: Signs and Symptoms
We see patients in our office who complain of these findings all the time, and only rarely are these people going to have PH. These are very nonspecific complaints and not surprisingly, because PAH remains a relatively uncommon disease, these are often confused with other conditions, and the diagnosis of PAH remains unacceptably delayed -- there is still an estimate of a 2-year delay from the time of symptom onset until a diagnosis is made. If we can raise our index of suspicion and proceed properly with the diagnostic evaluation, I believe that we can lessen the confusion with other conditions, and we can shorten the time to making a final diagnosis.

Slide 9. Assessment of PAH: Physical Examination
Physical examination turns out to be critical; we always hear this talked about, but I really think this is one disease state where physical exam is crucial. We need to be able to recognize the definitive physical exam findings that indicate pulmonary pressure overload, and subsequently, RV pressure overload; the presence of an enhancement of the pulmonic component of the second heart sound; the presence of an RV lift; signs of systemic venous congestion; or right-sided valvular insufficiency murmurs.
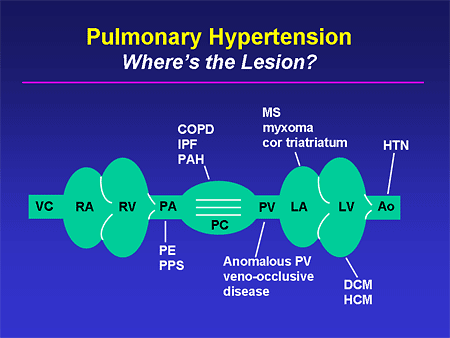
Slide 10. Pulmonary Hypertension: Where's the Lesion?
When we find these, I think it is very important that we take a moment to think anatomically and recognize that there are a whole host of conditions from the most mundane -- systemic hypertension -- through some of the more rare disorders that can either produce PH or can mimic it. If we take a second to think anatomically, we are going to be much better able to come up with the true diagnosis.
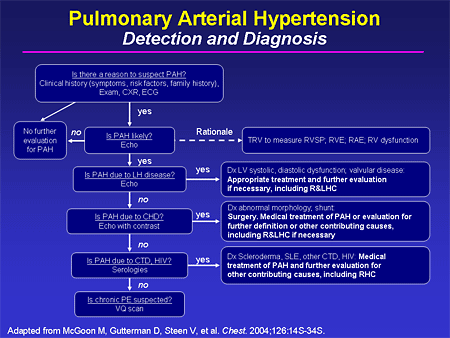
Slide 11. Pulmonary Arterial Hypertension: Detection and Diagnosis
In 2004, the American College of Chest Physicians (ACCP) published a supplement to Chest that is very worth your while to get and have it available. It is basically a soup-to-nuts treatment of PH, and in it is a very nice diagnostic algorithm -- this is only one half of it -- that can walk you through a reasonable diagnostic evaluation for a patient with suspected PH.

Slide 12. The Role of the Cardiologist in PAH
Informed interpretation of screening studies; I'm going to talk about 2 screening studies that we employ all the time. The first is the electrocardiogram (ECG) and the second is an echocardiogram (echo).
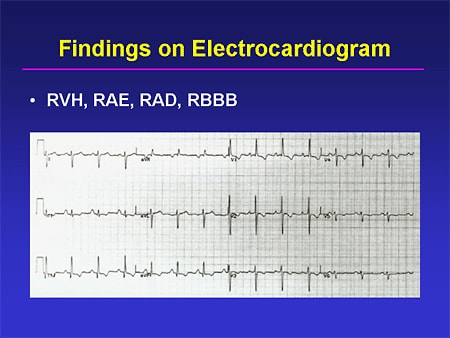
Slide 13. Findings on Electrocardiogram
An ECG can be very helpful here; ECG can show evidence of right-sided overload. You can see RV hypertrophy with very impressive positive forces in the right precordial leads; you can see right axis deviation; you may see the presence of P pulmonale or evidence of right atrial enlargement; and you can -- not in this particular tracing -- see either a full right bundle branch block, or an incomplete right bundle branch block, all of which will lend some supportive evidence to a diagnosis of PH. Of course, their absence does not rule out the disease, nor does their presence rule in the disease.

Slide 14. Assessment of PAH: Echocardiography
Echocardiography has clearly become the cornerstone and most important screening tool that we have for this. We all know that it can give us an estimate of the RV systolic pressure, but I want to emphasize that there is much more information that can be gotten from an echo as it relates to this disease state. What do the other right-sided chambers look like? What is their size? What is their function? Is there evidence of valvular disease on the right that can either be a primary issue for that patient or perhaps a secondary issue from the pulmonary pressure overload? Is there evidence of flow reversal in the inferior vena cava (IVC) that may go along with tricuspid insufficiency? Probably second most important to observations about right-sided chamber size and function is the lack of evidence of left-sided heart disease. If you see that, then I think you have to have your suspicion raised that this could be PAH.
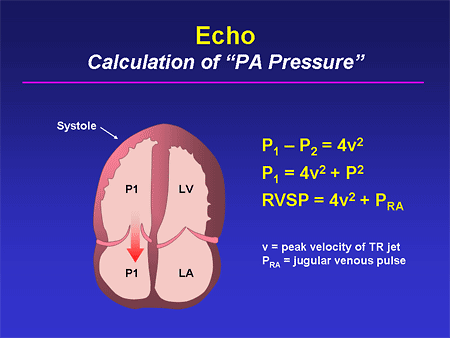
Slide 15. Echo: Calculation of "PA Pressure"
We must be able to recognize ourselves and communicate to our noncardiology colleagues what it means to get an estimated PA pressure from an echo. We need to be able to remind them that it requires the presence of a tricuspid insufficiency jet; it requires that the sonographer properly place the Doppler sample volume; it requires that the interpreter properly distinguish the peak velocity of that tricuspid insufficiency jet; and it requires that we make some reasonable assessment of the right atrial pressure. Of course, we can go wrong at all of those stages and we frequently hear, especially outside of cardiology, frustration over inaccuracies in the echo assessment of PA pressure. I think if we can gently communicate to our noncardiology colleagues that there are inherent potential flaws to this technique, it will help them understand some of these discrepancies a bit better.
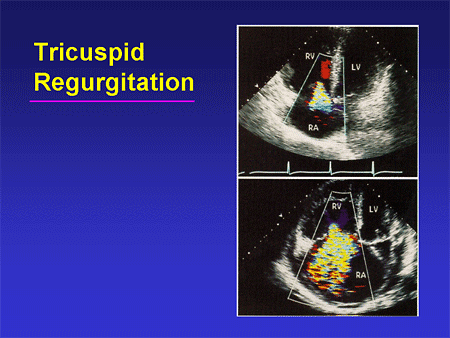
Slide 16. Tricuspid Regurgitation
Of course, the supportive evidence we might see can include tricuspid insufficiency, either mild amounts or more significant amounts, as well as chamber enlargement and chamber dysfunction.
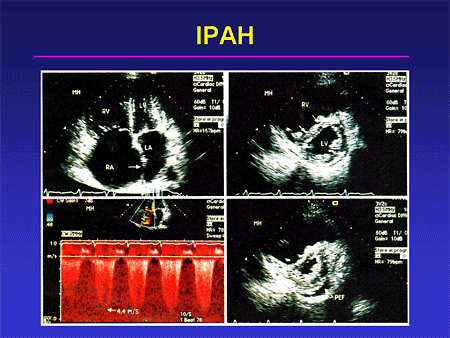
Slide 17. IPAH
A classic photo montage of a patient with idiopathic pulmonary arterial hypertension (IPAH) is shown here: the apical force showing very impressive right-sided chamber enlargement; the short axis view showing this septal shift; so-called D-shaped ventricle, both in diastole and in systole; and a tricuspid insufficiency jet with a very high velocity. This is a classic set of findings for somebody who has severe IPAH.

Slide 18. The Role of the Cardiologist in PAH
We must be willing to perform and interpret right heart catheterizations. I believe that virtually every single patient who is undergoing diagnostic evaluation for PH should have a hemodynamic study; an echo alone is not sufficient to make this diagnosis. Why do I say that?
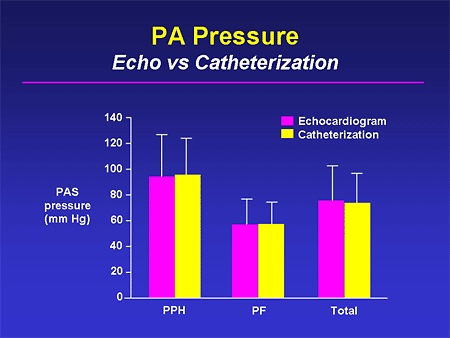
Slide 19. PA Pressure: Echo vs Catheterization
The first issue is accuracy. If you look at group mean data of the estimated PA pressure from an echo and compare it with that measured by catheterization, you have very nice agreement across groups. But the problem is the error bars are fairly substantial.
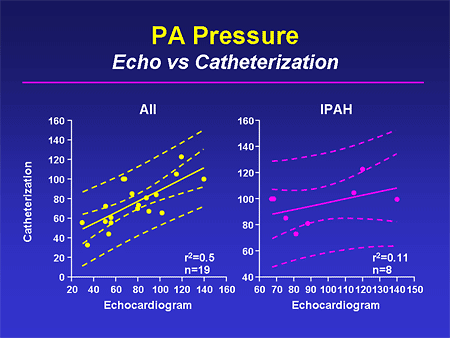
Slide 20. PA Pressure: Echo vs Catheterization
If instead, you look at individual data, and these have been published in the literature numerous times, you can see upwards of a 40% or 50% discrepancy between the echo estimate of the pressure and the directly measured pressure at right-heart catheterization. From an accuracy point of view, it is critical that the pressure is directly measured.

Slide 21. PHTN: Measurements
Two other values have to be gotten via catheterization. Wedge pressure; if you'll recall, the definition of this disease state includes the presence of a normal or near-normal pulmonary capillary wedge pressure, so it must be measured and it must be measured accurately. We know that in severe PH it can be difficult to obtain a good wedge pressure, but we must do everything in our power to get it and if we can't, we should consider whether we should more directly measure left-sided cardiac filling pressure. We use the wedge to generate a transpulmonary gradient, and this can be very helpful in some patients who seem to have their PH on the basis of left-sided heart disease or some other conditions.
The other factor that cannot be gotten invasively is cardiac output, and we can use this for 2 important reasons: The first is to get a sense of how much high cardiac output might be contributing to the pulmonary pressure we are measuring; and secondly, to help guide decisions about treatment.
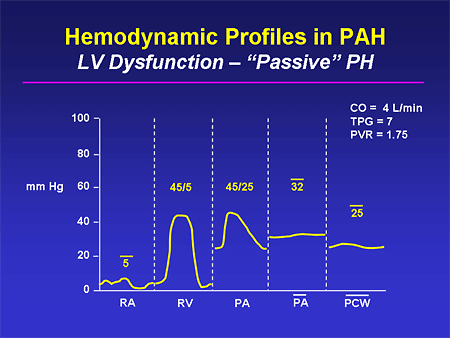
Slide 22. Hemodynamic Profiles in PAH: LV Dysfunction -- "Passive" PH
This is an example of a patient in whom you might be fooled by the echo. You can see that this patient has PH -- the mean PA pressure is in excess of 30 mm Hg. But simultaneously, the wedge pressure is high and the transpulmonary gradient, which you can either calculate or just visually estimate, is normal. And this is not somebody who has PAH; there is no lesion that exists at the level of the pulmonary artery here. It is instead passive from high left-sided cardiac filling pressure. And if this patient is treated for their heart failure, we will see a fall in their filling pressures and resolution of this PH in most cases.
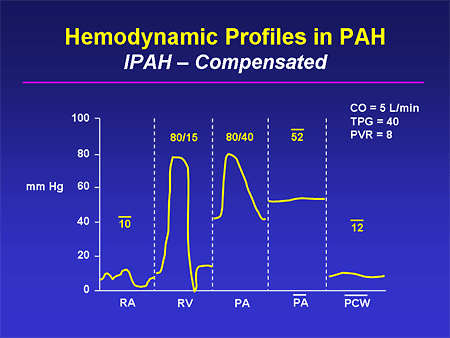
Slide 23. Hemodynamic Profiles in PAH: IPAH -- Compensated
Compensated IPAH might look like this, with very high PA pressures. The PA mean pressure obviously is grossly abnormal and the wedge pressure is normal. The transpulmonary gradient calculates to 40, but visually you can see how big it is; this is a classic finding in somebody who has IPAH. Notice that the patient is fairly well compensated; right-sided cardiac filling pressures are only modestly elevated; cardiac output is preserved.
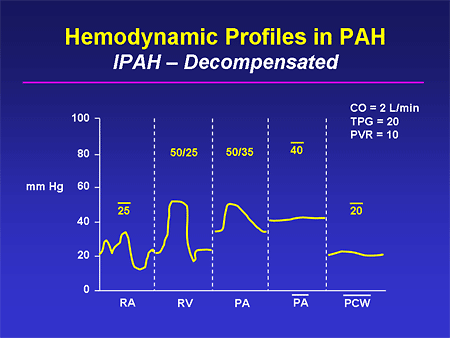
Slide 24. Hemodynamic Profiles in PAH: IPAH -- Decompensated
This represents a situation that can fool the physician. You have a patient with this disease, you have been treating them, they are clinically doing worse, and you do a follow-up echo and the estimate of the PA pressure has actually fallen; obviously that doesn't jibe. Well, the reason it has fallen is that this person is having decompensation of their RV function; right-sided cardiac filling pressures are quite high, cardiac output is quite low, and you can see that this person is basically in cardiogenic shock.
It also is an interesting phenomenon that you sometimes see an elevation in the wedge pressure in the late stages of this disease state. As the right ventricle fails progressively, there is more and more geometric abnormality of the left ventricle, and potentially underfilling of the left ventricle that can contribute a bit to this elevation in wedge pressure.
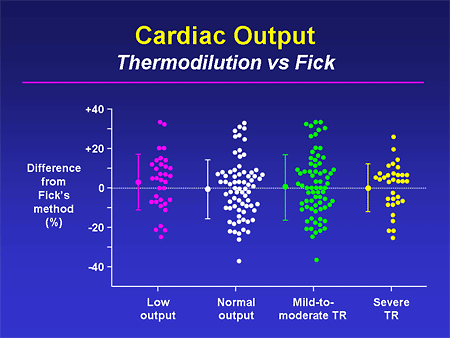
Slide 25. Cardiac Output: Thermodilution vs Fick
This is a plea to, if not actually calculate a Fick cardiac output, at least get a pulmonary artery oxygen saturation. Some people have described thermodilution cardiac outputs as random number generators; we all have seen numbers that are all over the place. This shows that regardless of what the underlying disease state is -- low output, high output, lots of tricuspid regurgitation (TR), not much TR -- you can see the huge amount of discrepancy between a Fick cardiac output and a thermodilution output. I believe that most of us feel more comfortable that the Fick is giving us the more accurate estimate of cardiac output.

Slide 26. The Role of the Cardiologist in PAH
Treatment of PAH commensurate with interest and ability.

Slide 27. Therapeutic Options for PAH
I'm not going to talk about treatment. This is a list of the things that one might consider as therapy; Dr. McLaughlin is going to spend her talk on this. I do want to emphasize that I think a decision must be made on a practitioner-to-practitioner basis about how involved in the treatment of this disease state you want to get. With oral therapies I think more people are becoming more comfortable treating this disease. But I think if you are not comfortable in treating more complex or more severe cases, you should at least have a good referral network so that you can get these patients into specialized centers.

Slide 28. The Role of the Cardiologist in PAH
Diagnosis and management of RV failure; this is a tricky one. As I think all of us know, we are relatively bad at understanding the right ventricle. We don't image it well; we don't model it well; and I think we tend to make our best guess based on clinical impression about whether a given patient's right ventricle is irreversibly failing or not. I do think that there is some hope on the horizon; a number of new diagnostic techniques are being evaluated for the assessment of RV function. I think we all have heard about magnetic resonance (MR) and how good MR can be for this.
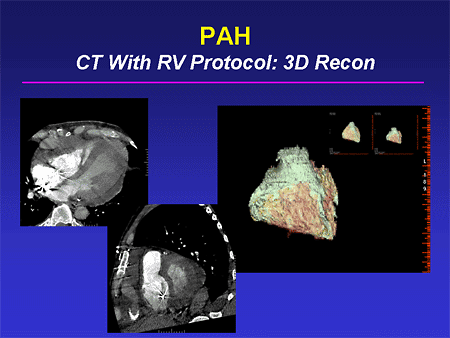
Slide 29. PAH: CT With RV Protocol: 3D Recon
We and other groups have been interested in the more widely applicable computed tomography (CT) scan, especially with newer CT technology, in terms of the scanners getting better, and also in terms of being able to deliver dye more accurately. By being able to gate scans in a better fashion, we can get very nice definition of RV size and function, even in the presence of intracardiac devices. (This patient has an implantable cardioverter defibrillator -- ICD.) And then we can do 3-dimensional reconstruction of these images and get very nice global, and we hope eventually regional, information about RV structure and function. These all are under investigation now and I think we will be seeing better techniques for RV assessment in the future.
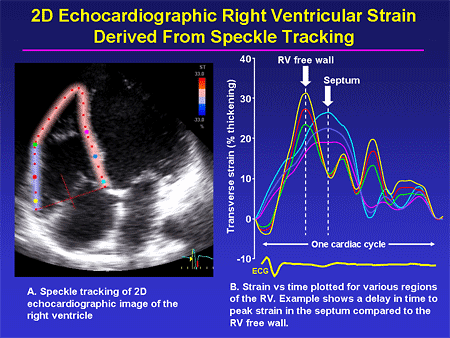
Slide 30. 2D Echocardiographic Right Ventricular Strain Derived From Speckle Tracking
Our echo colleagues also are doing some very interesting work in this field, especially looking at tissue Doppler and related techniques. This shows a newer technique called speckle tracking where we are trying to define whether the right ventricle becomes desynchronous; trying to define what parameters can be used to pinpoint those patients; trying to understand what relationship RV desynchrony may have to PAH. Perhaps we can intervene in much the same way that we do on the left side of the heart with resynchronization therapy, and improve RV function -- another new area of inquiry that I think we should all be keeping our eye on as we go through the next few

Slide 31. The Role of the Cardiologist in PAH: Conclusions
I'm going to close by reminding everybody that PAH is a cardiovascular disease. I believe that it is a disease state where patients are clearly optimally treated if cardiologists are actively involved. I hear too often out in the community pulmonologists who have been interested in this disease say, "I can't get any of my cardiologists interested; I can't get right heart catheterizations done." I think we need to reverse that; I think we need to be more actively involved. And if we can do that, I think we will see ongoing improvement in patient outcomes.

Slide 1. New Patient Population Research and Treatment Strategies in PAH

Slide 2. Outline
If you take nothing else away from this discussion, I would like to leave you with some of the points here. I'm going to emphasize the nontypical idiopathic pulmonary arterial hypertension (IPAH) patient. I'm going to tell you that despite what we have focused on in most of our trials (IPAH trying to be homogeneous), we are a very heterogeneous population of pulmonary arterial hypertension (PAH) patients. We have very different physiologies despite hearing that much of it is the same for our trials; in fact, we are very different in terms of how we get into the pathways for which we have therapies. Our comorbidities may be very different and because of that, when we think about therapies, I want us to focus on a way of getting into the different pathways. Our morbidities are different, the ways we break down drugs are different, and we are going to need randomized trials to see that therapies are safe and effective for particular populations, and I will emphasize one of the trials.
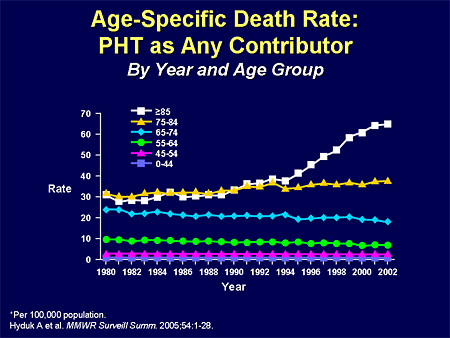
Slide 3. Age-Specific Death Rate: PHT as Any Contributor
Mensah from the Centers for Disease Control and Prevention (CDC) and the US Department of Health and Human Services, did a bunch of work for us in the pulmonary hypertension (PH) community in 2005. He and his group emphasized that if you look at the role of PH as a contributor to death in the United States, as a contributor to hospitalization, it is a huge increasing percentage relative risk. These are age-specific diagnoses in terms of relative risk. As you see, as we get older the relative percentage of patients who have PH as a contributor to death goes way up, and the numbers are incredible; 60 to 70 per 100,000 in the United States.
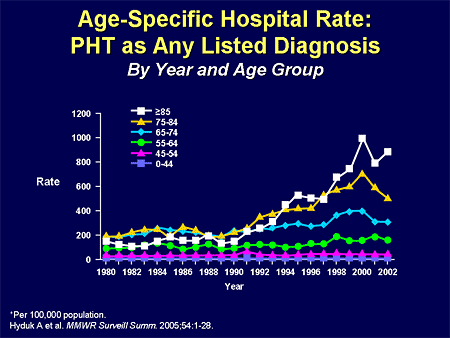
Slide 4. Age-Specific Hospital Rate: PHT as Any Listed Diagnosis
Look at this in terms of hospitalization. We are talking about nearly 1% of patients having PH related to their hospitalization, and in the older population, it isn't IPAH that is playing a role.

Slide 5. Our PAH Patients Are Not a Homogenous Population
We have increasing numbers of other forms of PAH and PH.
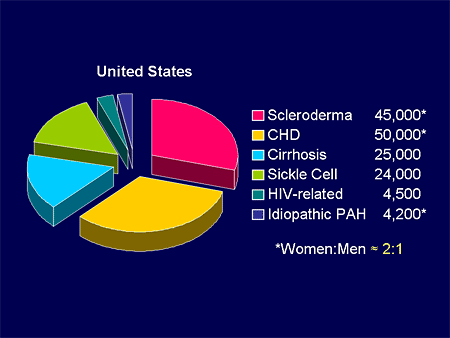
Slide 6.
If we look at this pie, we know that whether it's genetic forms and genetic triggers, whether it be sickle cell disease, cyanosis related to Eisenmenger syndrome, acquired forms of PH, rheumatologic disease, or various toxins, we live in a sea of relative triggers for PH; not just IPAH, but other potential causes.
If you look at that IPAH portion of the pie, it is a small percent of what we classify as PAH; maybe 3%. If you start including other potentials such as the toxin-related stuff, the non-IPAH disease, chronic obstructive pulmonary disease (COPD), we are talking about perhaps 150,000 to 200,000 patients with other forms of PH. We are talking about PAH; IPAH being maybe 1%-2% of this total pie.
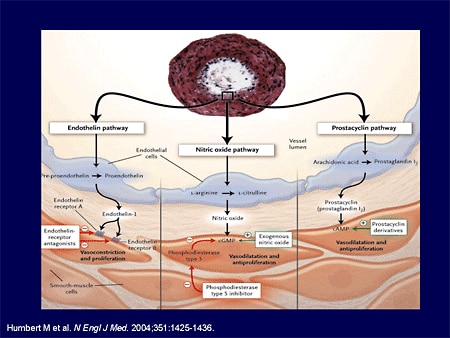
Slide 7.
When I began with PH, we had no idea in terms of the pathways and the triggers involved with PH. We are blessed in today's world to know not just major pathways, but the cellular and the matrix-based pathways and triggers of PH.
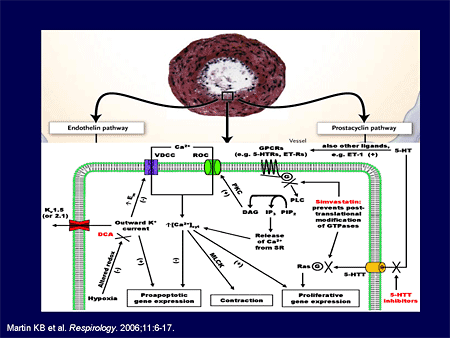
Slide 8.
And since we know this, we have a greater ability to understand mechanisms not just on the surface, the tip of the iceberg, but down under in terms of real disease pathophysiology. I'm going to give you some examples of this.
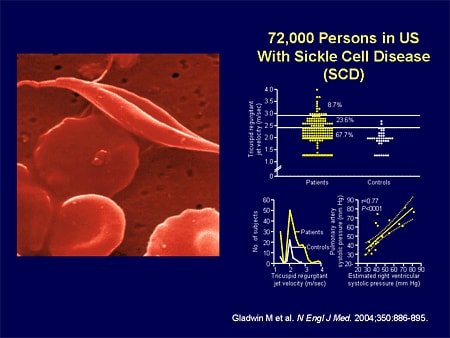
Slide 9. 72,000 Persons in US With Sickle Cell Disease (SCD)
Let's look at one form of PH; not typical IPAH but like PAH. Sickle cell disease; 70,000 people in this country have sickle cell disease; not a trivial number. Gladwin's group, about a year ago, showed us that in the sickle cell population, roughly a third of patients have echocardiogram (echo)-demonstrable PH.
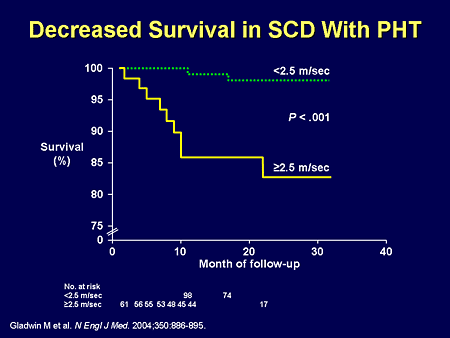
Slide 10. Decreased Survival in SCD With PHT
Dr. Mathier may say that echo may not be as accurate as catheterization, and we all agree with that, but there was one important finding based upon those echo findings: patients had significantly worse mortality in terms of survival. Echo was good enough as a screening tool to define a high-risk population, whether it be from the PH itself, or the other comorbid organ system disease. That's step 1. Step 2: is there a unique physiology involved with sickle cell disease-related PH? And the answer is absolutely.
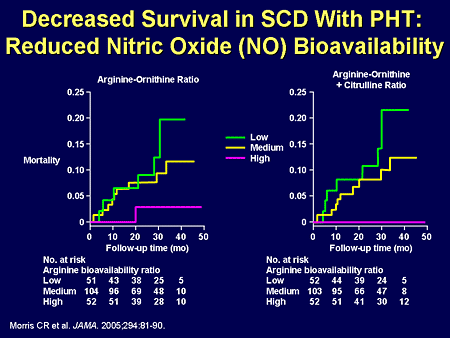
Slide 11. Decreased Survival in SCD With PHT: Reduced Nitric Oxide (NO) Bioavailability
We have learned that it may be a very specific entry into the nitric oxide pathway that happens with sickle cell disease -- here measured by various breakdown products along the nitric oxide synthase and arginase pathways.
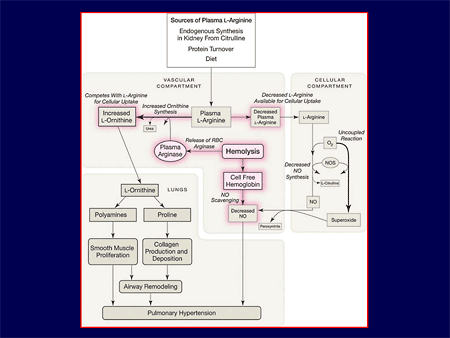
Slide 12.
We see here in a very complex scheme, but quite simple as well: whether you enter the scheme because of oxidative stress; whether you enter it at the top because of other multiple comorbid organ system issues in sickle cell; or you enter it by the typical issue with sickle cell, breakdown of red cells, releasing arginase, arginase is going to allow more arginine into the circulation, worsening contraction, and changing the nitric oxide availability throughout the cell or allowing cell-free hemoglobin to be out there and scavenge nitric oxide. You may know the nitric oxide pathway, but the targets for our therapy are going to be very different. Understanding the physiology is going to lead potentially, to a different therapy, and those therapies are going to interact at the very top here with other comorbid illness.
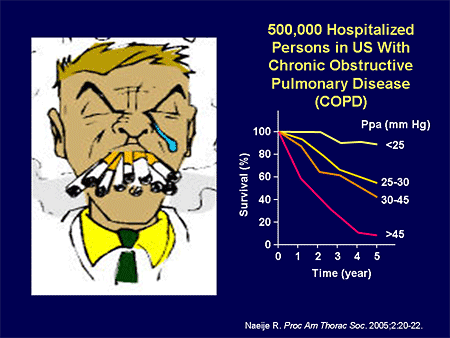
Slide 13. 500,000 Hospitalized Persons in US With Chronic Obstructive Pulmonary Disease (COPD)
What about COPD-related PH? This isn't trivial. As we said before, we have a huge number of patients who are hospitalized, and roughly one third of those patients have PH associated with them. And if you have PH, your survival is markedly decreased.
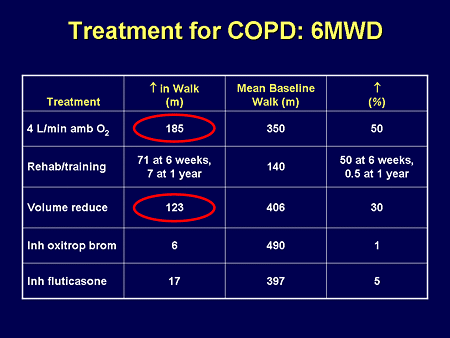
Slide 14. Treatment for COPD: 6MWD
We have therapies for COPD but they aren't very effective. That is why you haven't seen a lot of therapeutic trials, because COPD is not just a lung disease; it is a total body disease, and the ability to interact with morbidity and mortality in terms of having a real effect is difficult. And as you see, we have 2 therapies, in general, for COPD.
Slide 15. Direct Toxic Effects of Cigarette Smoke
Even knowing the various cascades in terms of how they affect PH, by entering into any of these different pathways, we remain very unsure that we will affect the critical determinant in terms of morbidity and mortality.

Slide 16. Populations Are NOT Identical
Despite a big group of PH patients, we are very different.
Slide 17.
Each one of us has very different entries into this system. Knowing this cascade may be very helpful. We may have an idea of a particular therapy, but at the end of the day, in the other forms of PH, we may have very early and premature aging of the rest of the body, and even though we may have a therapy that affects some of the outcomes measures, we may not be influencing the other determinants of survival.

Slide 18. Congenital Heart Disease: Prevalence
Let's focus for the last few moments on one of the fields that I love, which is congenital heart disease. Let me set the backdrop for you. Is it important or not in our world? It is the number one congenital birth defect in this country. In the United States, in the adult world, we estimate about 1 million patients have congenital heart disease. More adults than children have congenital heart disease; this affects us as adult caregivers.
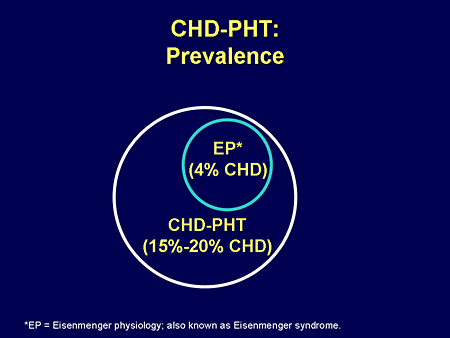
Slide 19. CHD-PHT: Prevalence
The prevalence of PH is not small; it is estimated that somewhere between 15% and 20% of that million with congenital heart disease have PH. And if you think about the worst form of PH, Eisenmenger physiology or Eisenmenger syndrome, it is about 4% of the total. Those are not small numbers, and significantly larger than even IPAH.
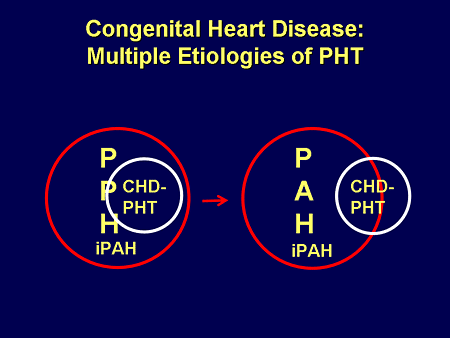
Slide 20. Congenital Heart Disease: Multiple Etiologies of PHT
We used to, for the sake of being included in the trials, consider ourselves identical to the rest of IPAH: we were within this same category, but we know that is not true. We know that a small cadre of that congenital heart disease-related PH is PAH, and a large percentage is outside of it.

Slide 21. Congenital Heart Disease: Multiple Etiologies of PHT
The PAH-related etiologies and triggers are those of flow and pressure; increased flow, left-to-right shunting causing eventual shunt reversal.

Slide 22. Congenital Heart Disease: Multiple Etiologies of PHT
But in fact, there are many other potential triggers in the same population; all the ones that you think about with secondary PH.
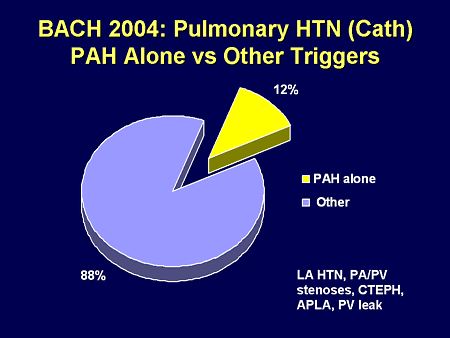
Slide 23. BACH 2004: Pulmonary HTN (Cath): PAH Alone vs Other Triggers
What do I mean? In our population in Boston, Massachusetts, in terms of our congenital heart disease-related PH, approximately 10% are related to PAH; the other 90% have other triggers for their PH, maybe left atrial hypertension.
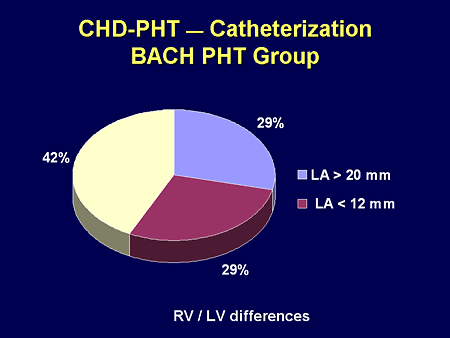
Slide 24. CHD-PHT -- Catheterization: BACH PHT Group
But the majority of our patients have elevated left atrial pressures despite being thought of as having typical Eisenmenger-derivative PH. They may get to left atrial hypertension because they may open shunts; it may be right-heart failure or left-heart failure; but the long and short of it is they have other contributors that may be hemodynamic in nature. Doing the catheterization is critical to that understanding.
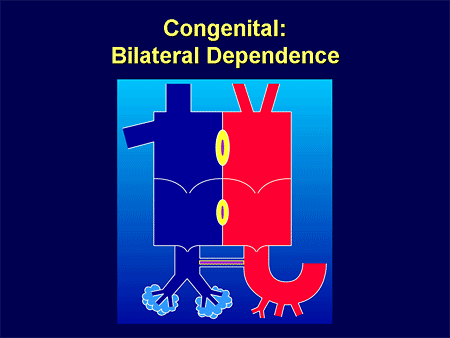
Slide 25. Congenital: Bilateral Dependence
My patients don't have normal conduit vessels; you think about PAH as an arteriolar disease.
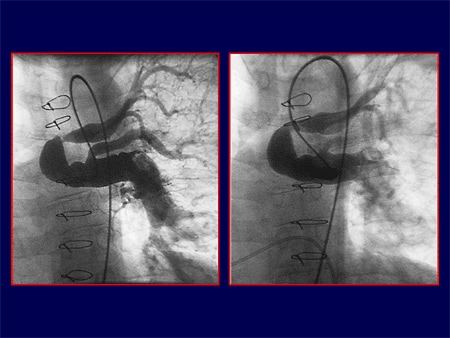
Slide 26.
These are their conduit major branch pulmonary arteries that are filled with stenoses and narrowings that may contribute to the resistance to the right ventricle. Whether we have a therapy for PAH or not, we don't know that they are going to affect these types of vessels. This is what we have to think about.
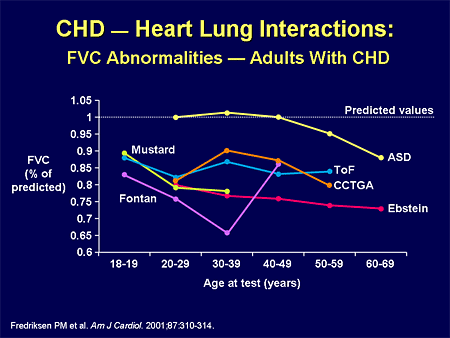
Slide 27. CHD -- Heart Lung Interactions: FVC Abnormalities -- Adults With CHD
When my patients have restrictive lung disease, they are cyanotic, and they have other hypoxia-mediated contributors to their PH.
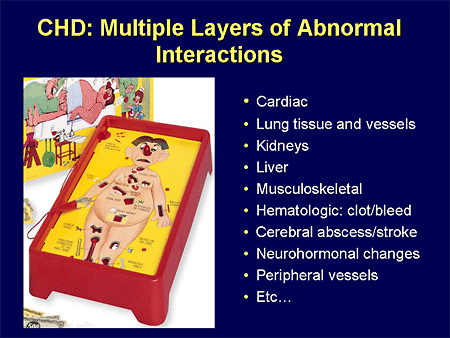
Slide 28. CHD: Multiple Layers of Abnormal Interactions
We have a whole host of other organ-system contributors that we have to think about when we have drug therapies that are metabolized through many of these same organs. I have to think about whether they are going to clear the drug in the same way.
Eisenmenger Physiology
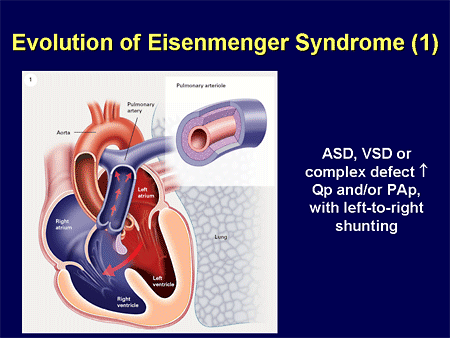
Slide 29. Evolution of Eisenmenger Syndrome (1)
Eisenmenger physiology; most of you are familiar with this concept; that whether it is a large hole in the atrium, the ventricles, or the great arteries, we are going to have left-to-right shunting.
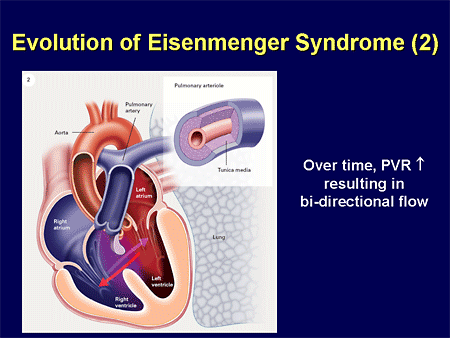
Slide 30. Evolution of Eisenmenger Syndrome (2)
That leads to an increase in either pulmonary blood flow or pulmonary pressure that is going to affect these pulmonary arteries and cause increased inflammation.
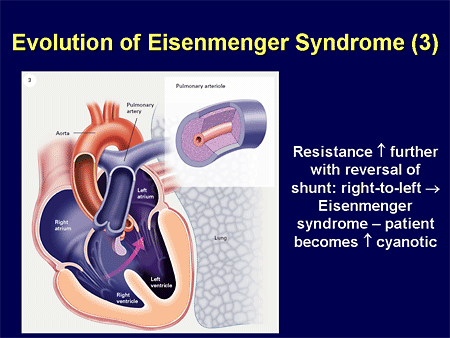
Slide 31. Evolution of Eisenmenger Syndrome (3)
This worsens pulmonary vascular resistance, ultimately leading to a reversal of that shunting, namely the Eisenmenger syndrome.

Slide 32. Symptomatology in Adults With EP
These patients are sick. They are blue, they are feeling bad, they have all the same symptoms of your PH patients from other etiologies, yet they look even worse. They come to you with chest pain, hemoptysis, cyanosis, and what is interesting, despite all of the therapies that you have heard about in congenital heart disease and PH, we have made no dent in the survival pattern of these patients at all.
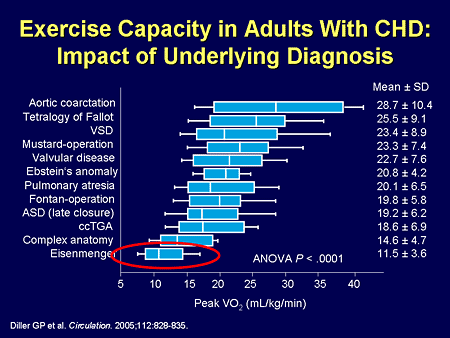
Slide 33. Exercise Capacity in Adults With CHD: Impact of Underlying Diagnosis
These patients with congenital heart disease are among our sickest. These are all the other congenital heart diseases that you have heard about; the lowest is Eisenmenger syndrome in terms of functional capacity as measured by peak oxygen consumption.
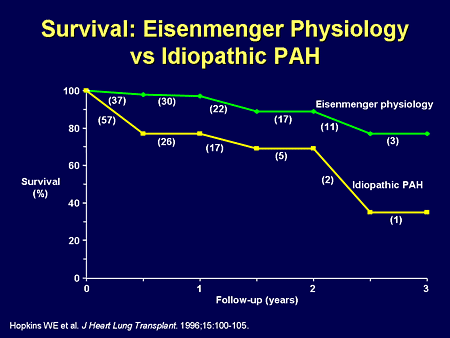
Slide 34. Survival: Eisenmenger Physiology vs Idiopathic PAH
And while survival may appear better than PAH, in the Eisenmenger physiology patient, survival is the worst of all of the congenital heart disease patients who we see.

Slide 35. Rationale
It is interesting; there is no evidence-based treatment for Eisenmenger physiology. Despite decades and decades of understanding these patients, we have no data-driven therapy for them. And like all of the rest of IPAH, we know that endothelin is implicated in the Eisenmenger syndrome. We know that bosentan, one of the dual endothelin receptor antagonists, has been shown to be effective in typical PAH patients, and because of that, there were a number of small open-label studies that suggested we may be able to show some benefit in these patients.

Slide 36. Rationale
That drove the Bosentan Therapy for Pulmonary Arterial Hypertension (BREATHE)-5 trial looking at the use of bosentan in Eisenmenger patients. We looked at whether or not patients had safety with this drug as measured by their oxygen saturations, improvement as measured by hemodynamic parameters, as well as 6-minute walk test.
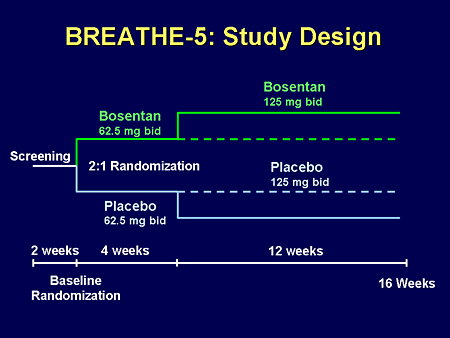
Slide 37. BREATHE-5: Study Design
It was a typical PAH-type protocol with a screening phase, a run-in phase of drug compared with placebo, and a longer phase of 3 months of full therapy, with various testing throughout the early and middle phases of the trial.

Slide 38. BREATHE 5: First RCT to Assess Therapy for EP
As you can imagine, because of all the potential triggers with PAH or PH in congenital heart disease, defining great inclusion and exclusion criteria was critical to the accuracy of this trial; it was an intense trial and we had tight reproducible specific endpoints.

Slide 39. BREATHE-5: Results
Let's review the results of the trial. By definition, there was a 2:1 randomization within the trial, so you see 37 patients randomized to bosentan with 17 patients receiving placebo. There was a female predominance, with 60% of patients being female. The typical age of inclusion was around 40 years of age, and this was a trial mostly of patients with ventricular septal defects compared with about one third of patients with atrial septal defects; typical for Eisenmenger syndrome.
These were sick patients; they had high pulmonary vascular resistance, and their mean pulmonary arterial pressures were approximately 70 to 75 mm Hg. Pulmonary blood flow was low, and by definition, QPQS, pulmonary to systemic blood flow was less than 1.0. Of interest, systemic vascular resistance was raised in this population, with an increase in systemic arterial pressure at baseline. There were no differences in baseline hemodynamics between the placebo and the bosentan group.
With regard to our first primary endpoint, we showed that therapy with bosentan over this relatively short-term trial did not worsen systemic pulse oximetry in our patients on bosentan. Of interest, compared with placebo, our patients did slightly better with a treatment effect that was noticeable. However, the trial was not powered to show this, and therefore, we can just say that our primary endpoint was reached. Patients were not worsened by bosentan therapy as assessed by pulse oximetry.
Having met the primary endpoint, we were able to look at our second primary endpoint, which was the effect of bosentan on pulmonary vascular resistance in these patients. What was remarkable to us, even in this short-term trial, was bosentan improved pulmonary vascular resistance compared with placebo significantly, with a treatment effect of nearly 500 dynes. This improvement in pulmonary vascular resistance was mostly secondary to a drop that was mild but real in pulmonary arterial pressure, with very little change in pulmonary blood flow. These results were confirmed in other analyses.
When we looked at other hemodynamic parameters, we saw that systemic vascular resistance dropped ever so slightly with bosentan, with a treatment effect compared with placebo that was nearly 800 dynes; this nearly reached significance. Again, the majority of that effect appeared to be due to a mild decrease in systemic arterial pressure, and less to systemic blood flow. That mild drop in mean systemic arterial pressure was not accompanied by any worsening in the patients, and there was no change in heart rate.
Now for the results that are of interest to most of you: how did our patients feel? How did they walk? How did they do? In fact, bosentan improved exercise capacity considerably, with a treatment effect of an improvement of 53 meters in 6-minute walking distance, and this was highly significant. Patients felt better; they reported an improvement in their functional class that was real and substantial, with a marked improvement, and an increase in patients in functional class II.
Patients on bosentan did not need to discontinue their therapy compared with placebo; in fact, there was significantly less discontinuation in the bosentan arms. In terms of serious adverse events, there were fewer serious adverse events, relatively speaking, compared with placebo.
In summary, I'm very pleased to present that in this first-ever randomized placebo-controlled study of ill Eisenmenger patients, even in a short-term therapy protocol, bosentan did not worsen our patients; it did not reduce systemic or oxygen saturation. At the same time, it significantly improved exercise capacity as measured by 6-minute walk distance. Physiologically, we have recognized that part of these effects is due to the significant reduction in pulmonary vascular resistance seen in these patients. The therapy with bosentan had a safety profile consistent with previous prospective clinical trials in other forms of PAH. In total, I'm very pleased to present for my co-investigators that the BREATHE-5 results suggest that bosentan is a new treatment option for patients with Eisenmenger syndrome who are severely ill.
Slide 40. Summary
Remember, there is a small percentage of the patients who we see in our practices who are classified as PAH of an idiopathic nature. We live in a world where the rest of those patients differ, and those are the patients we are going to have to face outside of the trials that you are going to hear about from Dr. McLaughlin. They have very different physiologies, they have premature aging. The drugs that we have been taught about don't necessarily have simple comorbidities associated with them in all the patients that we want to treat. We are looking forward to randomized trials in those specific patient populations, and you will begin to see those trials published more and more in the near future.

Slide 1. Applying Lessons of Heart Failure in PAH
We are a decade or two behind our colleagues on the left side of the heart; we have treatments now, but we still are learning how to apply them in combination and in clinical practice.
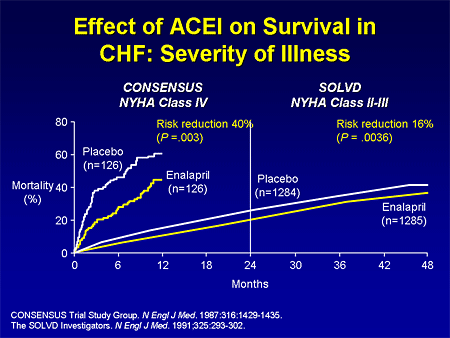
Slide 2. Effect of ACEI on Survival in CHF: Severity of Illness
Let's talk a little bit about what we have learned from heart failure trials and how that might apply to pulmonary hypertension trials, because we have lots of trials now that we need to apply to our clinical practice. One lesson we have learned is that the patient populations enrolling in clinical trials are different, and therefore some of the results that we are getting are different. I will remind you of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) trial in the mid 1980s. We were taking the sickest of the sick heart failure patients and randomizing them to angiotensin-converting enzyme (ACE) inhibitors or placebo. When you take really sick patients, you don't need very many patients to determine a survival benefit over a very short period of time; 250 patients gives us a survival benefit in CONSENSUS in a year.
And then as time goes by, we did not put those sick patients in randomized controlled trials; we were doing trials with the less ill patients. And when we look at patients with less advanced symptoms, functional class II and III as opposed to functional class IV, it takes a lot more patients, more than 2500 patients over 4 years, before we can find a survival benefit with an effective drug for heart failure -- ACE inhibitors.
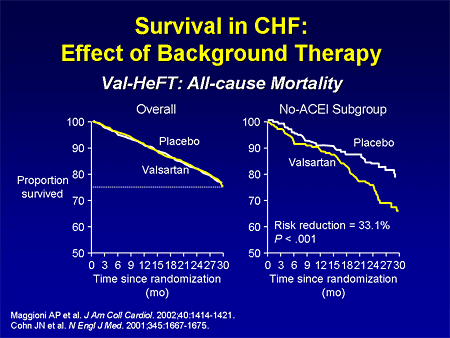
Slide 3. Survival in CHF: Effect of Background Therapy
We also saw the field change as we had more and more drugs approved for heart failure; more and more drugs that were demonstrated effective for heart failure because patients were being treated with those drugs. Then we were looking at newer drugs in heart failure; we were looking at patients treated with what we call add-on therapy, or step therapy.
If you look at the Valsartan Heart Failure Trial (Val-HeFT), looking at valsartan, when you look at the overall population, many of whom came in on effective therapies, we didn't see a treatment benefit. But if you took out the patients who were treated with ACE inhibitors, you saw the treatment benefit. So as patients come in on different therapies, it is harder to find a treatment benefit.
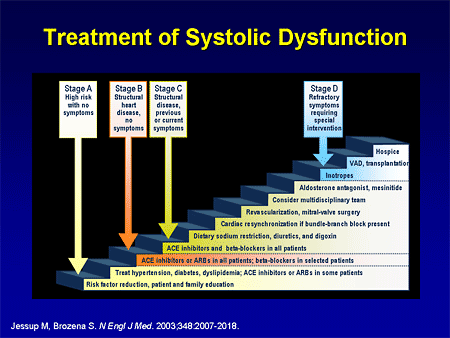
Slide 4. Treatment of Systolic Dysfunction
So now we have this very complex algorithm that we apply to treat heart failure -- to treat systolic dysfunction -- that we are starting to apply at an earlier and earlier stage of the disease. And this is exactly what is happening in pulmonary hypertension -- we are getting less ill patients in clinical trials; we are having more options for patients; and we are starting patients earlier.
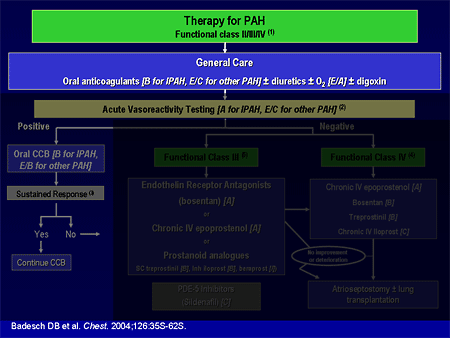
Slide 5. Therapy for PAH
I am going to go through the evidence-based algorithm that we have. Dr. Mathier, in the first presentation, alluded to these evidence-based guidelines that were published as a supplement to Chest almost 2 years ago. And Dr. Landzberg made the comment that maybe these guidelines don't apply to everybody, and he is correct. These are mostly for patients with pulmonary arterial hypertension (PAH), and most of these trials have excluded patients with complex congenital heart disease, sickle cell disease, chronic obstructive pulmonary disease (COPD), and some of the other areas that Dr. Mathier discussed.
Therapy for PAH includes some general care; we generally anticoagulate patients with idiopathic pulmonary arterial hypertension (IPAH) based on some observational survival studies. The data are less clear for other types of pulmonary hypertension, such as scleroderma-related or congenital heart disease. We use diuretics much the way that we do in left heart failure; we use oxygen to keep saturations greater than 90%, which we can do in most patients, perhaps except those unrepaired shunts that Dr. Landzberg just discussed. Digoxin has not been well studied, although it is generally used in patients with low cardiac output.
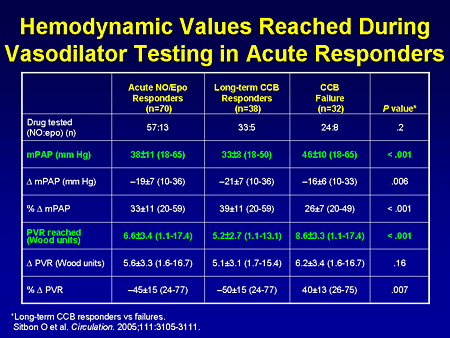
Slide 6. Hemodynamic Values Reached During Vasodilator Testing in Acute Responders
What really differentiated those patients who did well long-term is that the long-term responders had a reduction in their mean pulmonary artery pressure to about 33 mm Hg, and a reduction in their pulmonary vascular resistance to about 5.0. The patients who did not do well long-term, even though they had a reduction in mean pulmonary pressure of 20%, still had a mean pulmonary artery pressure in the 40s mm Hg, with a pulmonary vascular resistance of about 8.5.. That is a very brief summary of acute vasodilator testing, but the take-home point is that a very small proportion of patients with IPAH will respond.

Slide 7. Definition of a Responder
This is the new definition of a response: a fall in the mean pulmonary artery pressure by at least 10 mm Hg to an absolute mean pulmonary artery pressure, I believe, closer to 35 mm Hg, with an unchanged or an increased cardiac output. About 6% of IPAH patients will meet that response, and almost no patients with other types of PAH will meet that response. If a patient meets this response, they should be followed closely for the safety and efficacy of therapy. Calcium channel blockers are probably the most grossly overused therapy for pulmonary hypertension because they are ineffective in the vast majority of patients with PAH.
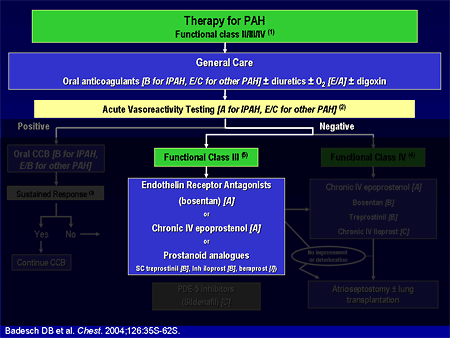
Slide 8. Therapy for PAH
That is a very small percentage of our patient population. Most of the patients we see these days have symptoms that are relatively advanced, and do not respond to acute vasodilators. There are now 5 US Food and Drug Administration (FDA)-approved therapies from 3 different classes for these patients. Dr. Landzberg mentioned the endothelin-receptor antagonists. Endothelin levels are high in patients with pulmonary hypertension, and blocking endothelin improves that disease. He mentioned the nitric oxide pathway, and we approach the nitric oxide pathway from the back end, by blocking phosphodiesterase with sildenafil. And then of course we can replace prostacyclin. Prostacyclin is deficient in pulmonary hypertension, and replacing it improves the outcomes.
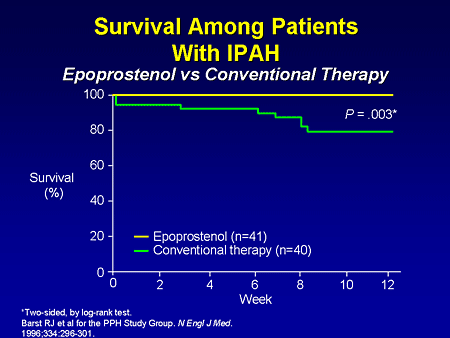
Slide 9. Survival Among Patients With IPAH: Epoprostenol vs Conventional Therapy
Let's go through some of the clinical trials. This is the sort of consensus of pulmonary hypertension; this is the first randomized controlled trial in pulmonary hypertension looking at replacing prostacyclin with intravenous (IV) epoprostenol. The sickest of the sick patients were enrolled in this trial, and after 12 weeks we demonstrate a mortality benefit; of 81 patients who died in this trial, all were randomized to conventional therapy. There also were improvements in 6-minute walk distance and hemodynamics.
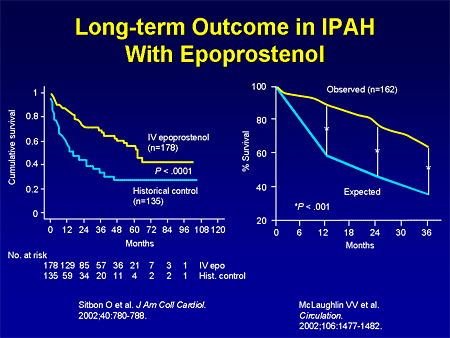
Slide 10. Long-term Outcome in IPAH With Epoprostenol
Longer-term data with IV epoprostenol also demonstrate improved survival, either compared with historical controls, or with what is expected based on the National Institutes of Health (NIH) equation. Of course, this is a very complex therapy; it is a continuous IV infusion, and while it is very effective, it is not for everyone.
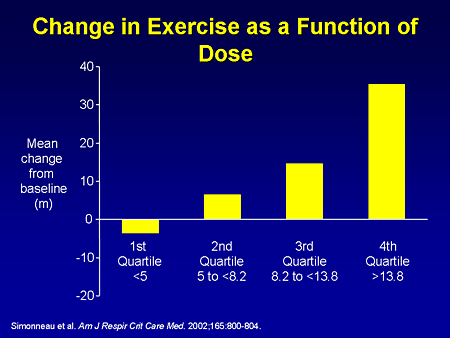
Slide 11. Change in Exercise as a Function of Dose
There are other ways to replace prostacyclin. The prostacyclin analogue treprostinil (Remodulin) was approved in the United States based on this study when it was evaluated subcutaneously in 470 patients with various types of pulmonary hypertension over 12 weeks, demonstrating an improvement in exercise tolerance that was clearly dose-related. If you gave adequate doses of subcutaneous treprostinil, there was a substantial improvement in 6-minute walk. This therapy can be limited by subcutaneous pain -- by pain at the site of the subcutaneous infusion -- and treprostinil can now also be used IV.
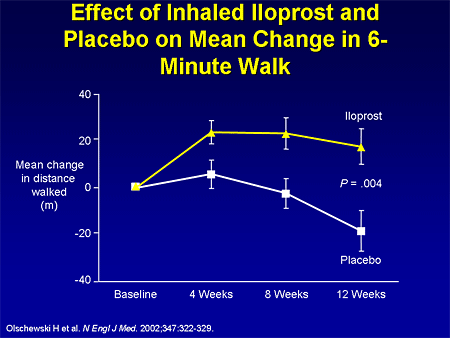
Slide 12. Effect of Inhaled Iloprost and Placebo on Mean Change in 6-Minute Walk
And then there is inhaled iloprost, an inhaled prostacyclin analogue that was approved in the United States based on a European study, the Aerosolized Iloprost Randomized (AIR) study, looking at inhaled iloprost 6 to 9 times per day vs placebo. This study looked at a composite endpoint, but I show you the 6-minute walk endpoint to compare apples with apples, and there was an improvement in 6-minute walk in the iloprost group compared with a reduction in the placebo group.
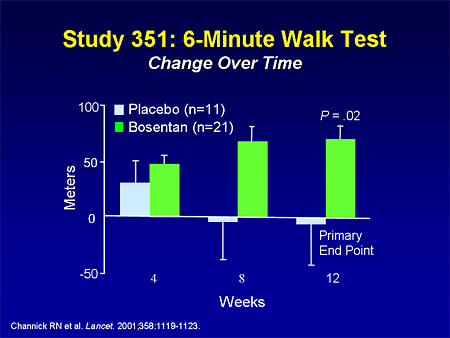
Slide 13. Study 351: 6-Minute Walk Test: Change Over Time
The endothelin receptor antagonists were the first oral therapy to come to the market. Bosentan is a dual endothelin receptor antagonist, and has been studied in 2 placebo-controlled trials; this one demonstrating an improvement of about 70 meters in the primary endpoint of 6-minute walk distance in patients with IPAH and PAH related to connective tissue disease.
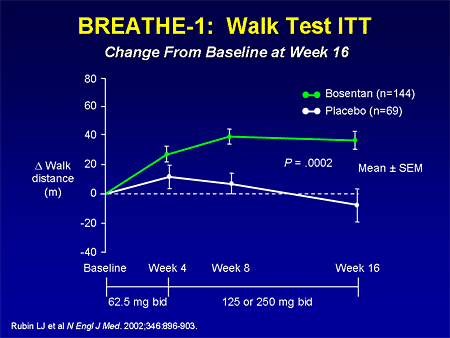
Slide 14. BREATHE-1: Walk Test ITT: Change From Baseline at Week 16
The larger Bosentan Therapy for Pulmonary Arterial Hypertension (BREATHE)-1 study demonstrated about a 40-meter improvement in 6-minute walk in 213 patients of the same entry criteria.
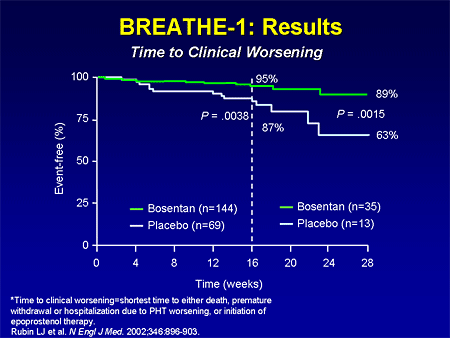
Slide 15. BREATHE-1: Results: Time to Clinical Worsening
The BREATHE-1 study also looked at an interesting secondary endpoint: time to clinical worsening. Patients improved in terms of survival with IV epoprostenol, and so we have less ill patients going into clinical trials. The other issue in pulmonary hypertension trials is that as patients get worse, we don't let them die; we put them on effective therapy. So this was a secondary endpoint that is like a composite endpoint of death, time to hospitalization and lung transplantation, or initiation of IV epoprostenol. At the end of 16 weeks, 5% of patients in the bosentan group met that endpoint, whereas 13% of patients in the placebo group met that endpoint. In a smaller number of patients who were followed in a blinded fashion up to 28 weeks out, those curves continued to diverge.
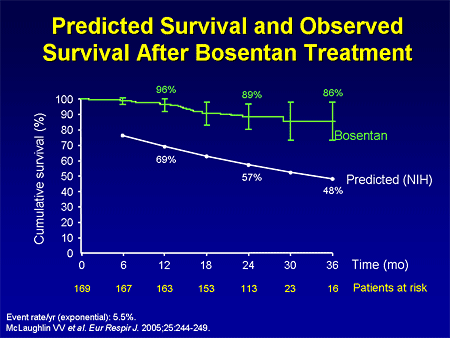
Slide 16. Predicted Survival and Observed Survival After Bosentan Treatment
We have observational evidence that first-line therapy with bosentan improves survival. This followed patients who were enrolled in either of the 2 clinical trials in the open-label extension. It demonstrated an improvement in survival with first-line bosentan therapy compared with what one would expect based on the NIH equation.

Slide 17. Bosentan and Liver Function Tests
Bosentan is an oral therapy; it obviously is easier than any of the prostacyclins that are currently FDA-approved. There are some cautions that I'd like to give you; about 11% of patients in the clinical trial had elevations of their transaminases greater than 3 times the upper limit of normal, and this needs to be monitored very carefully. One needs to check liver function tests on a monthly basis in patients treated with bosentan, and hemoglobin every 3 months because there is a small incidence of anemia.
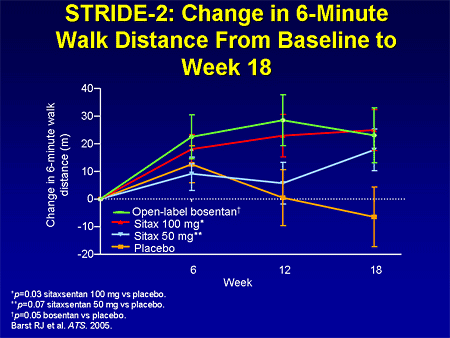
Slide 18. STRIDE-2: Change in 6-Minute Walk Distance From Baseline to Week 18
There are other endothelin receptor antagonists under clinical development. This is the primary endpoint from the Sitaxsentan To Relieve Impaired Exercise (STRIDE-2) in pulmonary arterial hypertension study, which is studying sitaxsentan. The dose of 100 mg achieved a statistically significant improvement, which was comparable with bosentan.
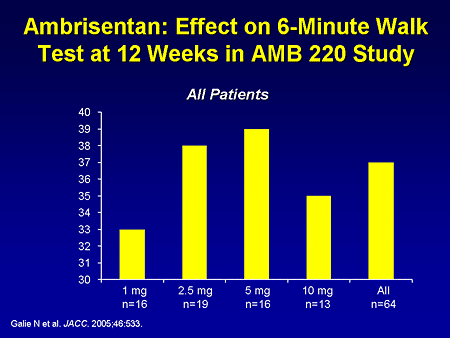
Slide 19. Ambrisentan: Effect on 6-Minute Walk Test at 12 Weeks in AMB 220 Study
There is another endothelin receptor antagonist under development, ambrisentan. These are the results from a phase 2 dose-ranging study demonstrating an improvement in 6-minute walk in this open-label study.
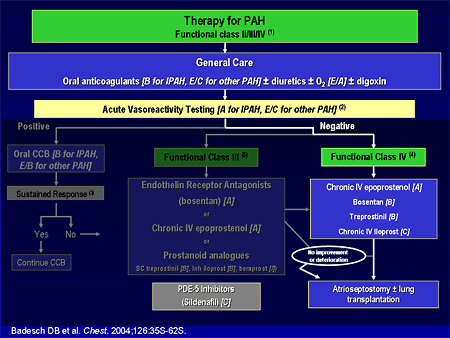
Slide 20. Therapy for PAH
As we go further on the algorithm, the very ill patients, the sickest of the sick, functional class IVs, intravenous epoprostenol gets the A grade because in the clinical trials, for most functional class IV patients, intravenous epoprostenol is the quickest, most effective therapy and should be used in those very ill patients.
At the time of the guidelines a year and a half ago, we left sildenafil floating because we did not have the results from the randomized clinical trial. When the guidelines are updated in the near future, sildenafil will move up, with a much stronger grade.
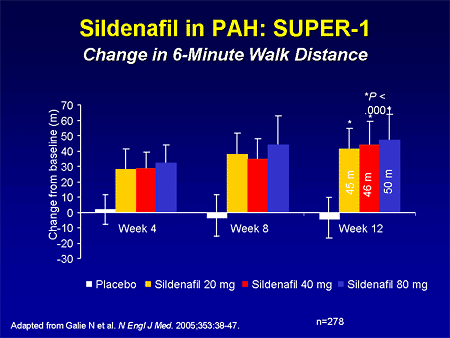
Slide 21. Sildenafil in PAH: SUPER-1: Change in 6-Minute Walk Distance
The trial that I'm speaking of is the Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) study, which was published in The New England Journal of Medicine recently. This looked at 278 patients with PAH who were idiopathic, related to either connective tissue disease, or repaired congenital heart disease. Patients were randomized to either placebo or 1 of 3 doses of sildenafil: 20, 40, or 80 mg, 3 times per day. The primary endpoint of 6-minute walk distance improved in all 3 groups by 45 to 50 meters, and there was not a dose response.
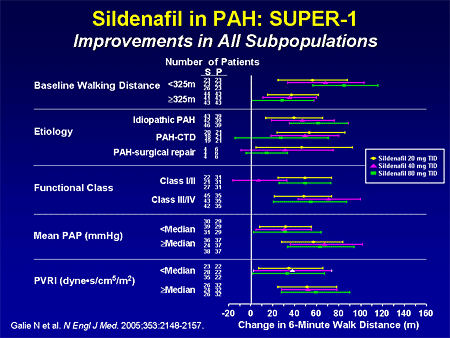
Slide 22. Sildenafil in PAH: SUPER-1: Improvements in All Subpopulations
There were also improvements in the different subgroups based on the 6-minute walk, above or below the median; all different types of pulmonary hypertension had improvements. Functional class I/ II and III/ IV had improvement.
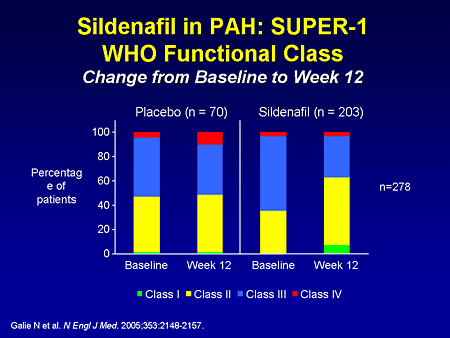
Slide 23. Sildenafil in PAH: SUPER-1: WHO Functional Class: Change From Baseline to Week 12
There were also improvements in functional class over the 12-week trial. In the placebo patients, functional class II and III, as you can see, are essentially unchanged at baseline and week 12, and in the sildenafil group, there were more patients in functional class II after 12 weeks, and less in functional class III.
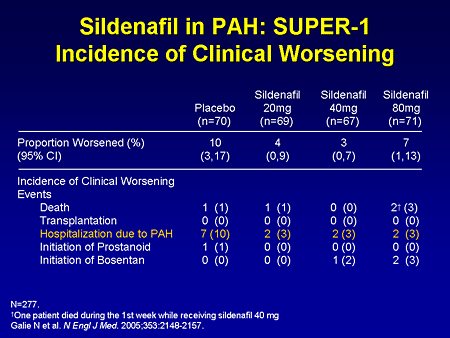
Slide 24. Sildenafil in PAH: SUPER-1: Incidence of Clinical Worsening
This is the composite endpoint of incidence of clinical worsening, which did not hit statistical significance at the end of 12 weeks, but you can see that overall there were less hospitalizations due to pulmonary hypertension.
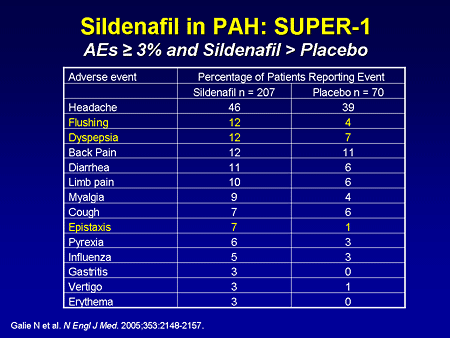
Slide 25. Sildenafil in PAH: SUPER-1: AEs Greater Than or Equal to 3% and Sildenafil Greater Than Placebo
This drug seems to be relatively well tolerated; the most common side effects included flushing and dyspepsia. Some patients complained of heartburn, particularly the first day or two of therapy. The other side effect that I'd like to point out that was a little more common in the sildenafil group was epistaxis.
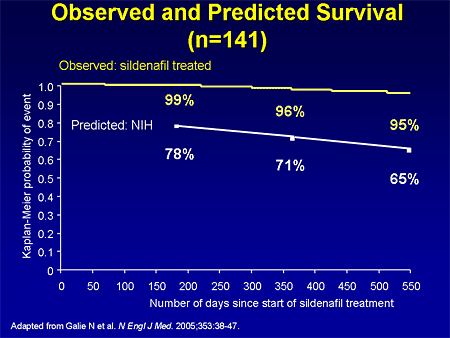
Slide 26. Observed and Predicted Survival (n=141)
In the long-term extension trial in patients who were titrated up to the 80 mg dose, there was also an improvement in survival compared with what one would expect based on the NIH equation. With that trial, sildenafil was approved in the United States at a dose of 20 mg, 3 times per day.
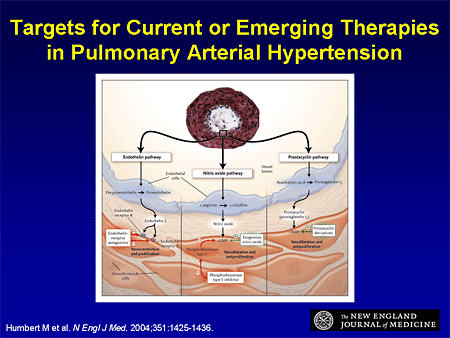
Slide 27. Targets for Current or Emerging Therapies in Pulmonary Arterial Hypertension
The other lesson we learned from heart failure is, now that we have therapies from 3 different classes that are effective, what about starting to combine these therapies? Dr. Landzberg showed you this slide that depicts the endothelin pathway; we have effective therapy here. The nitric oxide pathway -- we have effective therapy coming in the back end, blocking phosphodiesterase; and the prostacyclin pathway -- we have several effective prostacyclins to treat pulmonary hypertension. So combination therapy is going to be the next step.
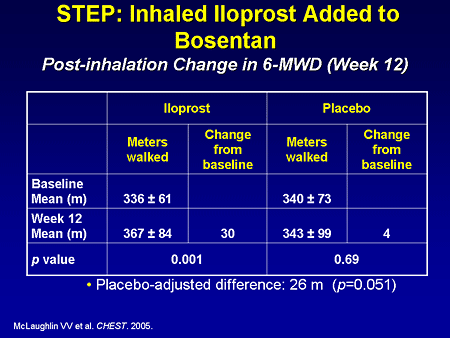
Slide 28. STEP: Inhaled Iloprost Added to Bosentan: Post-inhalation Change in 6-MWD (Week 12)
There are a few data available; the Iloprost Inhalation Solution Safety and Pilot Efficacy Trial in Combination with Bosentan for Evaluation in Pulmonary Arterial Hypertension (STEP) trial looked at patients who were on bosentan, adding iloprost vs placebo, and demonstrated a 26-meter placebo-adjusted difference in the small pilot trial.
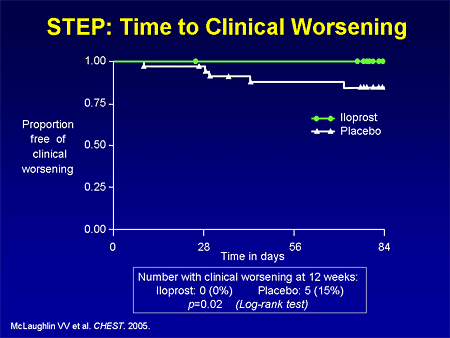
Slide 29. STEP: Time to Clinical Worsening
They also hit a time-to-clinical-worsening endpoint.

Slide 30. COMPASS Program
The first of many combination trials and one that I'd like to highlight is the COMPASS trial because this makes imminent sense; this is combining the 2 oral agents, which is obviously attractive for patients. This is a very unique trial. This is where we should go in pulmonary hypertension -- where we've been in left heart failure -- a true endpoint trial. We have lots of drugs that can improve 6-minute walk distance, but the bigger question for us as cardiologists and for our patients is, how do we affect long-term outcomes? How do we affect the morbidity and mortality of this disease?
The COMPASS trial is a large trial looking at patients on sildenafil randomized to bosentan or placebo, with the primary endpoint of morbidity and mortality; clinically relevant endpoints. This is also going to include a pharmacoeconomic analysis, which I think is very important in the current era.
Slide 31. Combination Therapy Trials
There are many other combination therapy trials that are either in progress or will be started soon, so this is really where we have been in heart failure, and where we are going in pulmonary hypertension.

Slide 32. Other Potential Investigational Therapies
We don't have a cure yet, but we have 3 good options. We still have more work to do, and there are other novel therapies that are going to be hopefully investigated in the coming years.
So we are learning lessons from heart failure. We have drugs that target different pathways, and we are going to start soon combining those pathways. Our profile of patients in clinical trials has changed, and we are going to need larger clinical trials to demonstrate benefits, especially as we use add-on therapy. But I think we are aligned in the pulmonary hypertension community, and I'm very hopeful that we will soon have some of these large trials, similar to what we have seen in heart failure.




/https%3A%2F%2Fassets.over-blog.com%2Ft%2Fcedistic%2Fcamera.png)
/http%3A%2F%2Fwww.gnmhealthcare.com%2Fclients%2Fimages%2F12%2F07%2F28%2F2450011_0.gif)
/http%3A%2F%2Fanmisims.canalblog.com%2Fimages%2FLIERRE_fleur445.gif)
/https%3A%2F%2Fassets.over-blog.com%2Ft%2Fcedistic%2Fcamera.png)