| Maladie de Gaucher de type 1 : anomalie de l’adhésion plaquettaire et risque de saignement muqueux |
Les patients qui présentent une maladie de Gaucher de type 1 peuvent avoir une tendance clinique significative au saignement, sans rapport avec le niveau de leur taux de plaquettes. Pour vérifier le rôle joué par les plaquettes une étude israélienne a inclus 48 patients avec une maladie de Gaucher de type 1, 52 porteurs (parents d’enfants avec une maladie de Gaucher) et 19 témoins bien portants. Chez les patients l’adhésion des plaquettes était significativement plus faible que chez les témoins ou chez les porteurs. L’adhésion plaquettaire n’était pas modifiée par la prise d’une enzymothérapie substitutive spécifique de la maladie, mais elle était améliorée après splénectomie. Cette diminution de l’adhésion était liée à un défaut plaquettaire intrinsèque. Un saignement muqueux était signalé chez 17 patients (35,4%) et était associé à une anomalie de l’adhésion (odd ratios : 5,73). Chez 5 patients il existait une baisse de l’agrégation plaquettaire et tous présentaient une diminution de l’adhésion plaquettaire. Le défaut d’agrégation plaquettaire n’était pas associé avec des saignements muqueux. Une anomalie de l’adhésion plaquettaire constitue une thrombocytopathie majeure chez les patients qui présentent une maladie de Gaucher de type 1 et peut expliquer en partie la tendance augmentée au saignement.
Platelet adhesion defect in type I Gaucher Disease is associated with a risk of mucosal bleeding
[27-04-2011]
Galia Spectre 1, Batia Roth 1, Galia Ronen 3, Dror Rosengarten 4, Deborah Elstein 4, Ari Zimran 4, David Varon 1, Shoshana Revel‐Vilk 2
1 Coagulation Unit
2 Paediatric Haematology/Oncology Department, Hadassah‐Hebrew University Medical Centre, Jerusalem, Israel
3 Radiology Department, Sourasky Medical Centre, Tel Aviv
4 Gaucher Clinic, Shaare Zedek Medical Centre, Jerusalem, Israel
*Correspondence:G. Spectre, Coagulation Unit, Hadassah‐Hebrew University Medical Centre, Jerusalem 91120, Israel.
E‐mail: galias@hadassah.org.il
Summary
Patients with type I Gaucher Disease (GD) may have a clinically significant bleeding tendency that is disproportionate to their platelet count. We hypothesized that impaired platelet adhesion might contribute to bleeding tendency. Adult patients with type I GD with platelet counts ≥130 × 109/l and haematocrit ≥30% (n = 48), obligatory carriers (n = 52), and healthy controls (n = 19) were studied. Platelet adhesion, using the IMPACT‐R (Cone and Plate(let) Analyser), and platelet aggregation were determined. Type I GD patients had significantly lower platelet adhesion [surface coverage %, median (interquartile range)] 4·6 (3·2–7·5), compared to controls, 8·7 (7·6–10·3), or carriers, 8·1 (6·5–9·4; P = 0·001). Platelet adhesion was not affected by the use of disease‐specific enzyme replacement therapy but was improved in patients after splenectomy, 7·2 (5·8–9·3). Mixing tests showed that the reduced adhesion was an intrinsic platelet defect. Mucosal bleeding was reported in 17 (35·4%) patients and was associated with abnormal adhesion [P = 0·037, with an Odds Ratio (95% confidence interval) of 5·73 (1·1–29·6)]. Five patients (22%) had reduced platelet aggregation, all of whom had reduced platelet adhesion. Platelet aggregation defect was not associated with mucosal bleeding. In conclusion, platelet adhesion defect is a major thrombocytopathy in type I GD patients and can explain part of the increased tendency to bleeding.
A bleeding tendency is a prominent feature of type I Gaucher disease (GD). The main bleeding symptom of these patients is mucocutaneous, which manifests as epistaxis, gingival bleeding, easy bruising, and/or heavy menstrual bleeding (Granovsky‐Grisaru , 1995; Larsen , 2003; Zimran , 2005). Increased bleeding tendency in GD is commonly attributed to thrombocytopenia secondary to hypersplenism and/or to bone marrow infiltration. Reduction in the activity of various coagulation factors, in particular low factor XI levels, and increased fibrinolysis, have also been described, mainly in unsplenectomized patients (Hollak , 1997; Katz , 1999; Deghady , 2006; Giona , 2006). A high gene frequency of both factor XI deficiency and type I GD in Ashkenazi Jews can explain the relatively common concurrence of both genetic disorders (Seligsohn , 1976; Berrebi , 1992). However, it has been considered that some GD patients have a clinically significant bleeding tendency that is not proportional to their platelet counts or coagulation abnormalities. Thus, an additional cause for bleeding may be an abnormal platelet function (Kelsey , 1994; Gillis , 1999; Giona , 2006). Abnormal platelet aggregation has been described in seven patients with type I GD in a series of 32 patients (22%) from our clinic (Gillis , 1999) and in six of 15 patients (40%) in another study (Giona , 2006).
Adhesion of platelets to the injured vessel wall, the initial step of the haemostatic response, is crucial for normal platelet function. To date, this important aspect of platelet response has not been studied in GD. We hypothesized that impaired platelet adhesion might contribute to the bleeding diathesis observed in patients with GD. In the present study we tested platelet adhesion under arterial shear conditions in patients with type I GD, obligatory carriers of the disease, and healthy volunteers. We used the IMPACT‐R [Cone and Plate(let) Analyser] technique (Shenkman , 2000). In addition, mixing studies were performed in order to evaluate the differential role of platelets, plasma, and red blood cells (RBCs) in platelet adhesion in GD.
Materials and methods
Patients and controls
Type I GD adult patients followed at the Gaucher Clinic, Shaare Zedek Medical Centre were eligible for this study. Diagnosis of GD was made by demonstration of decreased glucocerebrosidase activity in leucocytes (Beutler & Kuhl, 1970) and by mutation analysis at the DNA level (Beutler , 1992). In a few patients bone marrow findings of Gaucher cells in bone marrow, with a combination of two mutated alleles were acceptable in the absence of an enzymatic diagnosis. Lower platelet counts and/or haematocrit levels are known to be associated with abnormal adhesion using the IMPACT‐R system (Varon , 1997; Kenet , 1998), therefore we included only patients with platelet counts ≥130 × 109/l and haematocrit ≥30%. Data were recorded from the clinic medical files of all patients, including year of birth, ethnic origin, mutation analysis, history of bleeding episodes, history of splenectomy, and history of medical treatment, i.e. enzyme replacement therapy (ERT) or substrate reduction therapy (SRT). Disease severity was calculated by the severity score index (SSI; Zimran , 1989). Obligatory carriers, parents of children with GD coming to the clinic at the time of study period, and a group of hospital personnel as healthy controls were also enrolled in the study.
All study participants signed an informed consent form. The study was approved by the local ethics (Helsinki) committee.
Blood counts and platelet adhesion tests
Samples for complete blood count were collected in EDTA tubes. Platelet adhesion under flow conditions was studied using the IMPACT‐R (Cone and Plate (let) Analyser; DiaMed, Cressier, Switzerland; Shenkman , 2000). Blood samples for the IMPACT‐R test were collected in citrate tubes containing 0·38% sodium citrate, and were analysed within 3 h after blood draw. Citrated whole blood (130 μl) were placed on polystyrene plates (Nunc, Roskilde, Denmark) and subjected to a shear rate of (1800/s) for 2 min using a rotating teflon cone. The wells were then thoroughly washed with tap water, stained with May–Gruenwald stain (Sigma, Rehovot, Israel). The image of adhered platelets was analysed with an inverted light microscope (Olympus, Tokyo, Japan) connected to an image analysis system (Galai, Migdal Haemek, Israel). Two parameters of platelet adhesion were evaluated: percent surface coverage (SC, %) which is the percentage of total area covered by platelets (single platelets and clusters/aggregates) and the average size (AS, μm2) of the polystyrene bound platelet clusters/aggregates. The samples were analysed in duplicates, and the higher platelet adhesion result was used for statistical analysis.
The normal values of the IMPACT‐R test were previously specified as SC: ≥7% and AS: ≥23 μm2, based on the 5th percentile of log transformed data from 98 adult controls (Revel‐Vilk , 2009).
Preparation of platelet rich plasma, platelet poor plasma, and red blood cells
Platelet rich plasma (PRP) was prepared by centrifugation of anti‐coagulated whole blood 180 g for 12 min at room temperature. Red blood cells were isolated by further centrifugation of the remaining layer of whole blood at 1200 g for 5 min. The supernatant consisted of platelet poor plasma (PPP).
Mixing studies
Mixing studies were performed in nine pairs of patients and healthy volunteers, matched for blood type. Normal RBC (adjusted to the haematocrit of 40%) were reconstituted with patient’s PRP, incubated for 5 min with gentle rotation (10 rpm) and then analysed by the IMPACT‐R test. In the same manner, normal PRP was reconstituted with the patients’ RBC, and patients’ PPP was added to normal whole blood. In all experiments, autologous reconstitution of the healthy volunteer’s blood served as a control.
Platelet aggregometry
Platelet aggregation was measured by a routine platelet aggregometer (PACKS‐4, Helena Laboratories, Beaumont, TX, USA) using the following agonists (obtained from Diamed, Switzerland): ADP (10 μmol/l), epinephrine (10 μmol/l), and collagen (5 μg/ml). Platelet aggregation was considered normal if maximal aggregation amplitude was >60%.
Statistical analysis
Characteristics of study subjects are presented as nominal data. The percentages are presented with Fisher’s exact 95% confidence interval (CI). Nominal variables are presented as median (interquartile range) or mean (95% CI), where applicable.
Differences in age, platelet count, haematocrit, and SC/AS measurements between the different groups were assessed using the parametric and non‐parametric tests for normally and non‐normally distributed data, respectively. Adjustment of P values to correct for possible significance resulting from performance of multiple tests on the same data (Bonferroni like correction‐Hommel adjusted p; Wright, 1992) was performed with WINPEPI (PEPI‐for‐Windows, Version 2.8, March 2007).
Differences for mixing tests were evaluated by a paired T‐test.
To compare between GD patients with and without mucosal bleeding a logistic regression model was used. The following variables were considered: platelet count, haematocrit, SC/AS measurement, platelet aggregation test, SSI, and ERT.
Statistical analysis was performed with the Statistical Package for the Social Sciences (SPSS), version 14 for Windows. A P value <0·05 was considered significant.
Results
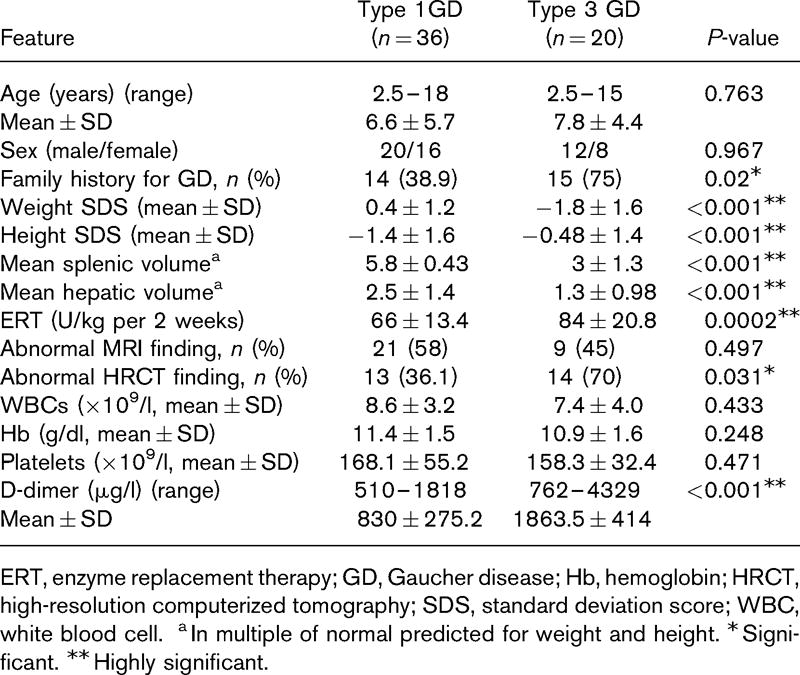
Forty‐eight adult patients with type I GD, 52 obligatory carriers, and 19 controls were enrolled to this study. The clinical characteristics of the GD patients are presented in Table I.
I Characteristics of patients with type I Gaucher disease.
| | No | % |
|---|
| Total patients |
48 |
|
| Male |
23 |
48 |
| Ashkenazi Jewish origin |
47 |
98 |
| Mutations |
| N370S homozygous |
23 |
48 |
| Compound heterozygous (N370S/other) |
25 |
52 |
| Severity Score Index (SSI) |
| Mild (1–10) |
31 |
65 |
| Moderate (11–25) |
16 |
33 |
| Severe (26–30) |
1 |
2 |
| Splenectomy (total or partial) |
17 |
35 |
| Medical therapy |
36 |
75 |
 Table 1 Table 1
Effect of gaucher disease on platelet adhesion
Patients with type I GD had a significantly lower SC as compared to controls and obligatory carriers (Table II). The platelet counts of GD patients were not significantly different from controls; however they were significantly lower as compared to obligatory carriers. No differences in AS measurements were found between patients, obligatory carries and controls.
II Laboratory characteristics of patients and obligatory carriers and healthy volunteers.
| | Patients | Controls | Carriers | P‐value* | P‐value† | P‐value‡ |
|---|
| Number (M:F) |
48 (23:25) |
19 (13:6) |
52 (26:26) |
– |
|
– |
| Platelet count, ×109/l |
193 (153–252) |
222 (200–245) |
249 (217–302) |
0·06 |
0·003 |
0·06 |
| Haematocrit, % |
40·7 (37·3–44·1) |
43 (40·5–45·8) |
41·1 (37·2–45) |
0·18 |
0·52 |
0·31 |
| Surface coverage, % |
4·6 (3·2–7·5) |
8·7 (7. 6–10·3) |
8·1 (6·5–9·4) |
0·002 |
0·002 |
0·36 |
| Average size, μm2 |
24·1 (20·1–33·9) |
28·0 (24·0–34·0) |
27·0 (24·0–37·0) |
0·15 |
0·07 |
0·86 |
Data presented as median (interquartile range). M, male; F, female.
*P value for Gaucher patients compared to control.
†P value for Gaucher patients compared to obligatory carriers.
‡P value for controls compared to obligatory carriers.
Table 2
Given that differences in platelet counts could potentially affect the difference observed in SC, 15 participants with platelet counts <160 × 109/l were excluded from the analysis in order to attain a median platelet count that was not significantly different between patients, obligatory carriers, and controls (P = 0·1). Also in this subgroups analysis, SC was significantly lower in patients, median (interquartile range), 5·9 (3·4–9), as compared to obligatory carriers, 8·1 (6·7–9·4) or controls, 8·8 (7·9–10·2; P = 0·001).
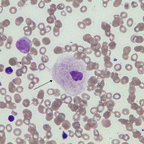
Correlation was found between SC and platelet counts in patients with type I GD (correlation coefficient 0·34, P = 0·017, Spearman correlation test). No correlation was found with age and the SSI. No correlation was found between SC and platelet count in controls or in obligatory carriers. Representative photographs of platelet adhesion in a patient and a healthy control are shown (Fig 1).

Figure 1 Reduced platelet adhesion in Gaucher disease. A representative platelet adhesion picture (IMPACT‐R, Cone and platelet analyser) of a healthy control (right): Surface coverage (SC) 9% Average size (AS) 27 μm2, and a patient with Type I Gaucher disease (left) SC 3·3%, AS 23 μm2.
Effect of ert and splenectomy on platelet adhesion
ERT‐treated patients had similar platelet counts, SC, and AS of aggregates compared to untreated patients (Table III). Splenectomized patients on ERT had significantly higher platelet counts and SC as compared to patients on ERT only. Only one splenectomized patient did not receive ERT and was excluded from this analysis.
III Characteristics of patients with Gaucher type I with or without enzyme replacement therapy (ERT).
| | No ERT | ERT only | ERT & splenectomy* | P‐value† | P‐value‡ | P‐value§ |
|---|
| Number |
11 |
20 |
16 |
– |
– |
– |
| Age, years |
39 (35–48) |
40 (25–59) |
48·5 (37–61) |
0·82 |
0·16 |
0·12 |
| Platelet count, ×109/l |
154 (138–204) |
169 (142–193) |
262 (214–302) |
0·99 |
0·002 |
<0·001 |
| Haematocrit, % |
40·6 (38·8–42·4) |
39·7 (36·6–42·9) |
41·3 (39·1–43·6) |
0·44 |
0·89 |
0·58 |
| Surface coverage, % |
3·8 (1·9–7·5) |
3·8 (2·4–5·3) |
7·2 (5·8–9·3) |
0·99 |
0·09 |
0·002 |
| Average size, μm2 |
20·0 (17·1–25·1) |
25·6 (22·5–35·8) |
24·9 (22·3–31·7) |
0·21 |
0·16 |
0·79 |
| Mucosal bleeding |
6 (54%) |
7 (35%) |
4 (25%) |
0·29 |
0·15 |
0·7 |
Data presented as median (interquartile range). ERT, enzyme replacement therapy.
*One splenectomized patient who did not receive ERT was excluded from the analysis.
†P value for patients without therapy compared to those who received ERT.
‡P value for patients without therapy compared to those who received ERT and underwent splenectomy.
§P value for patients who received ERT compared to who received ERT and underwent splenectomy.
Table 3
Although a trend of higher platelet count was found in splenectomized patients compared to controls (Fig 2A), the SC of those patients tended to be lower compared to controls (Fig 2B).
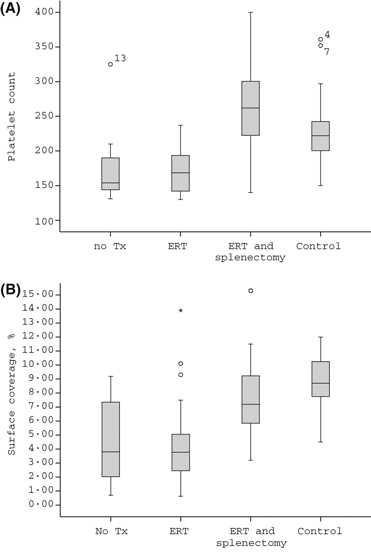
Figure 2 (A) Platelet counts in patients with Gaucher disease and controls. A box plot for the platelet count (×109/l) in untreated patients (no Rx, n = 11), patients receiving only ERT (ERT = 20), splenectomized patients also receiving ERT (ERT and splenectomy, n = 16), and controls (n = 19). A trend for higher platelet count in splenectomized patients on ERT compared to controls (P = 0·066). (B) Surface coverage in patients with Gaucher disease and controls. A box plot curve for the surface coverage (% IMPACT‐R) in untreated patients (no Rx, n = 11), patients receiving only ERT (ERT n = 20), splenectomized patients receiving also ERT (ERT and splenectomy, n = 16) and controls (n = 19). A trend for lower surface coverage in splenectomized patients on ERT as compared to controls (P = 0·071).
No correlation was found between platelet counts and SC during analysis of the different treatment groups, i.e. untreated with intact spleen, ERT with intact spleen, and ERT with splenectomy.
Bleeding history in patients with gaucher disease
Twenty‐eight patients reported a previous history of bleeding episodes (58·3%, 95% CI 43·2%‐72·4%). Eleven patients (22·9%) reported only easy bruising. Mucosal bleeding was reported 17 patients (35·4%), including epistaxis, menorrhagia, gingival bleeding, rectal bleeding, and prolonged bleeding after cuts or surgery. A bleeding history was not obtained in one patient. A history of mucosal bleeding was not associated with the SSI, ERT, platelet counts, haematocrit level, and/or SC/AS measurements.
To improve the diagnostic value of the IMPACT‐R test as a potential predicative test for mucosal bleeding, the results of both the SC and the AS measurements were used to define an abnormal test. An abnormal IMPACT‐R test, (defined as an abnormal SC < 7% and/or AS < 23 μm2) was found in 32 patients (66·7%, 95% CI 51·6–79·6%) and was associated with history of mucosal bleeding. An abnormal IMPACT‐R test was found in 15/17 (88·2%, 95% CI 63·6–98·5%) of patients with mucosal bleeding as compared to 17/30 (56·6%, 95% CI 37·4–74·5%) of patients without mucosal bleeding (P = 0·037). The Odds Ratio (95% CI) for mucosal bleeding in GD patients with an abnormal IMPACT‐R test was 5·73 (1·1–29·6).
Mixing studies in patients and controls
Platelet adhesion in the IMPACT‐R system is influenced by platelets, RBCs, and plasma components (Shenkman , 2000; Peerschke , 2007). To test the differential role of each component in the observed reduced platelet adhesion in GD, we performed mixing studies in nine pairs of patients and controls. Patient PRP mixed with control RBC resulted in a lower SC compared to control PRP mixed with control RBC, mean (95% CI), 3·5% (1·5–5·5%) and 8·5% (7·2–9·8%), respectively (P < 0·001; Fig 3).
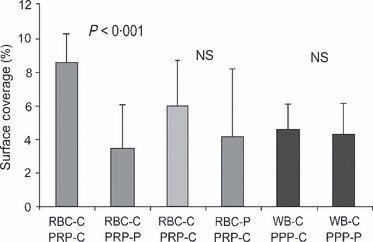
Figure 3 Mixing studies in patients and controls. RBC, red blood cell; PRP, platelet rich plasma; PPP, platelet poor plasma; WB, whole blood; C, control; P, patient; NS, not significant. Surface coverage (%) of reconstituted samples of patients and controls (n = 9) as described in material and methods. Surface coverage was significantly reduced when RBC‐C were reconstituted with PRP‐P. No change in SC was observed when patient PPP or RBC was reconstituted with control blood.
Mixing of control PPP with control whole blood diluted the sample and reduced platelet adhesion, mean SC (95% CI), 4·54% (3·34–5·74%). However, mixing of patient PPP with control whole blood did not reduce platelet adhesion further: mean SC (95% CI), 4·29% (2·85–5·72%). Mixing of patient RBC with control PRP did not change platelet adhesion (for the whole group of patients, n = 8), mean SC (95% CI), 5·98% (3·7–8·26%; Fig 3). However, a splenectomized patients’ RBC mixed with normal PRP reduced SC as compared to autologous mixing of control RBC and control PRP (n = 5, P = 0·04). This phenomenon did not occur in the patients with intact spleens (n = 3, P = 0·4).
Platelet aggregation
Platelet aggregation in response to epinephrine, ADP, collagen and ristocetin was tested in 22 patients. Platelet counts, SC/AS measurements, and rate of abnormal IMPACT‐R test were not different in patients who were tested or not for platelet aggregation. Isolated abnormal platelet aggregation in response to epinephrine was observed in three (13%) patients (Kambayashi , 1996). Abnormal platelet aggregation in response to ADP and/or collagen was observed in five (22%) patients, all of whom had an abnormal IMPACT‐R test [positive predictive value of 100% (95% CI 89–100%)]. Aggregation in response to ristocetin was normal in all patients. Platelet aggregation defect alone was not associated with mucosal bleeding (P = 0·32).
Discussion
This study was designed to assess the platelet adhesion in patients with type I GD. Platelet adhesion was found to be lower in patients with GD compared to obligatory carriers and controls. Platelet adhesion was not affected by the use of ERT and was improved after splenectomy. A platelet adhesion defect, measured by the IMPACT‐R system, occurred in two‐thirds of GD patients and was associated with history of mucosal bleeding.
The IMPACT‐R system measures both the adhesion of platelets to the polystyrene surface (SC), as well as the aggregation of circulating platelets around adherent platelets associated with the release reaction (AS). The significantly lower SC in GD patients compared to controls suggests that a platelet adhesion defect is a major thrombocytopathy in patients with GD. This thrombocytopathy might be missed when testing platelet function only with the platelet aggregometer. Indeed, abnormal platelet aggregation was found in only one‐fifth of the patients in this study and others (Gillis , 1999; Giona , 2006).
The mechanism of reduced platelet adhesion in patients with type I GD is not clear. Based on mixing tests, reduced adhesion is an intrinsic platelets defect and not affected by RBCs or plasma of these patients. The increased plasma levels of glucocerebroside in patients with GD may affect platelet activation (Nilsson , 1982; Aerts & Hollak, 1997; Gousset , 2002). However, this mechanism could possibly explain platelet abnormalities only in untreated patients. Alternatively, extracts of platelets were found to be rich in an aggregated, activated form of the glucocerebrosidase (Yatziv , 1974), thus lack of the enzyme in GD might cause increased glucocerebroside in the platelets and affect platelet function.
In our study, patients receiving ERT did not have higher platelet counts or platelet adhesion compared to untreated patients. This is in contrast to previous studies where platelet counts and platelet aggregation were shown to be improved after starting ERT (Hollak , 1997; Gillis , 1999; Giona , 2006). By excluding patients with low platelet counts, we might have influenced the chance to find differences in platelet counts and platelet adhesion between ERT‐treated and untreated patients.
In contrast, splenectomy was associated both with improved platelet counts and platelet adhesion as measured by the IMPACT‐R system. The improved platelet adhesion found after splenectomy might be directly related to the higher platelet counts. Alternatively, splenomegaly has been suggested to cause chronic platelet activation that might lead to platelet exhaustion with reduced function (Humphries & Hess, 1994; Hollak , 1997).
The study inclusion criterion of a platelet count >130 × 109/l has resulted in a relatively high percentage of splenectomized patients (35%) in the era of disease‐specific ERT.
The trend to lower SC in splenectomized GD patients compared to controls, in the presence of a tendency for higher platelet counts suggests that the platelet adhesion defect we observed in GD patients is a true pathological finding and is not only a reflection of the low platelet counts. The observation of a lower SC in a subgroup analysis of patients with platelet counts >160 × 109/l, supports a similar conclusion.
Preliminary data from mixing studies indicate that RBC from GD patients with intact spleens may reduce the SC of normal platelets, as compared to RBC from splenectomized patients. These preliminary results suggest that the better platelet adhesion profile of splenectomized patients could also result from elimination of a yet unknown effect of splenomegaly on RBC physiology under high shear stress. Further research is needed to study the mechanism of the platelet adhesion defect in GD patients.
Of interest is the association of abnormal platelet function measured by the IMPACT‐R system with history of mucocutaneous bleeding. No similar association was reported with the more commonly performed platelet aggregation test. Studying whole blood platelet adhesion under high shear rate may be a more relevant parameter for correlation with bleeding symptoms. In previous studies, the IMPACT‐R method was shown to be effective in the assessment of platelet function in thrombocytopenic patients (Kenet , 1998) and in patients undergoing cardiac surgery (Gerrah , 2004). Recently, the IMPACT‐R method was shown to be a useful screening tool for bleeding disorders in children evaluated for bleeding tendency (Revel‐Vilk , 2009).
The diagnosis of bleeding risk is clinically important in preparing patients before surgery or delivery. The current recommendations in patients with GD include performing a complete history of bleeding tendency (including easy bruising and recurrent gum or nose bleeds, and heavy menstrual bleeds), a complete blood count, coagulation factors assays if the prothrombin time and/or partial thromboplastin time are abnormal, platelet aggregation tests, and von Willebrand factor levels and activity (Ioscovich , 2005; Zimran , 2005). Platelet adhesion studies are currently used in the Gaucher Clinic, Shaare Zedek Medical Centre, as part of the routine check of the haemostatic system before surgical procedures or delivery.
In conclusion, platelet adhesion defect is commonly found in type I GD patients and can explain some of the increased bleeding tendency in these patients.
Testing platelet adhesion should be considered before surgery, delivery or dental procedures in order to assess the need for platelet transfusions, antifibrinolytic agents or desmopressin (DDAVP) in the individual patient.
The first two authors contributed equally to the paper.
References
 (Exportez format texte tabulé) (Exportez format texte tabulé)
• Aerts, J.M. & Hollak, C.E. (1997) Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clinical Haematology, 10, 691–709.
• Berrebi, A., Malnick, S.D., Vorst, E.J. & Stein, D. (1992) High incidence of factor XI deficiency in Gaucher’s disease. American Journal of Hematology, 40, 153.
• Beutler, E. & Kuhl, W. (1970) Detection of the defect of Gaucher’s disease and its carrier state in peripheral‐blood leucocytes. Lancet, 1, 612–613.
• Beutler, E., Gelbart, T., Kuhl, W., Zimran, A. & West, C. (1992) Mutations in Jewish patients with Gaucher disease. Blood, 79, 1662–1666.
• Deghady, A., Marzouk, I., El‐Shayeb, A. & Wali, Y. (2006) Coagulation abnormalities in type 1 Gaucher disease in children. Pediatric Hematology and Oncology, 23, 411–417.
• Gerrah, R., Snir, E., Brill, A. & Varon, D. (2004) Platelet function changes as monitored by cone and plate(let) analyzer during beating heart surgery. The Heart Surgery Forum, 7, E191–E195.
• Gillis, S., Hyam, E., Abrahamov, A., Elstein, D. & Zimran, A. (1999) Platelet function abnormalities in Gaucher disease patients. American Journal of Hematology, 61, 103–106.
• Giona, F., Palumbo, G., Amendola, A., Santoro, C. & Mazzuconi, M.G. (2006) Platelet function and coagulation abnormalities in type 1 Gaucher disease patients: effects of enzyme replacement therapy (ERT). Journal of Thrombosis and Haemostasis, 4, 1831–1833.
• Gousset, K., Wolkers, W.F., Tsvetkova, N.M., Oliver, A.E., Field, C.L., Walker, N.J., Crowe, J.H. & Tablin, F. (2002) Evidence for a physiological role for membrane rafts in human platelets. Journal of Cellular Physiology, 190, 117–128.
• Granovsky‐Grisaru, S., Aboulafia, Y., Diamant, Y.Z., Horowitz, M., Abrahamov, A. & Zimran, A. (1995) Gynecologic and obstetric aspects of Gaucher’s disease: a survey of 53 patients. American Journal of Obstetrics and Gynecology, 172, 1284–1290.
• Hollak, C.E., Levi, M., Berends, F., Aerts, J.M. & van Oers, M.H. (1997) Coagulation abnormalities in type 1 Gaucher disease are due to low‐grade activation and can be partly restored by enzyme supplementation therapy. British Journal of Haematology, 96, 470–476.
• Humphries, J.E. & Hess, C.E. (1994) Gaucher’s disease and acquired coagulopathy. American Journal of Hematology, 45, 347–348.
• Ioscovich, A., Briskin, A., Abrahamov, A., Halpern, S., Zimran, A. & Elstein, D. (2005) Uncomplicated outcome after anesthesia for pediatric patients with Gaucher disease. Canadian Journal of Anaesthesia, 52, 845–847.
• Kambayashi, J., Shinoki, N., Nakamura, T., Ariyoshi, H., Kawasaki, T., Sakon, M. & Monden, M. (1996) Prevalence of impaired responsiveness to epinephrine in platelets among Japanese. Thrombosis Research, 81, 85–90.
• Katz, K., Tamary, H., Lahav, J., Soudry, M. & Cohen, I.J. (1999) Increased operative bleeding during orthopaedic surgery in patients with type I Gaucher disease and bone involvement. Bulletin/Hospital for Joint Diseases, 58, 188–190.
• Kelsey, H., Christopoulos, C., Gray, A.A. & Machin, S.J. (1994) Acquired pseudo‐pseudo Bernard‐Soulier syndrome complicating Gaucher’s disease. Journal of Clinical Pathology, 47, 162–165.
• Kenet, G., Lubetsky, A., Shenkman, B., Tamarin, I., Dardik, R., Rechavi, G., Barzilai, A., Martinowitz, U., Savion, N. & Varon, D. (1998) Cone and platelet analyser (CPA): a new test for the prediction of bleeding among thrombocytopenic patients. British Journal of Haematology, 101, 255–259.
• Larsen, E.C., Connolly, S.A. & Rosenberg, A.E. (2003) Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 20‐2003. A nine‐year‐old girl with hepatosplenomegaly and pain in the thigh. New England Journal of Medicine, 348, 2669–2677.
• Nilsson, O., Hakansson, G., Dreborg, S., Groth, C.G. & Svennerholm, L. (1982) Increased cerebroside concentration in plasma and erythrocytes in Gaucher disease: significant differences between type I and type III. Clinical Genetics, 22, 274–279.
• Peerschke, E.I., Silver, R.T., Weksler, B.B., Yin, W., Bernhardt, B. & Varon, D. (2007) Examination of platelet function in whole blood under dynamic flow conditions with the cone and plate(let) analyzer: effect of erythrocytosis and thrombocytosis. American Journal of Clinical Pathology, 127, 422–428.
• Revel‐Vilk, S., Varon, D., Shai, E., Agmon, Y., Hyam, E., Daas, N., Miskin, H. & Weintraub, M. (2009) Evaluation of children with a suspected bleeding disorder applying the Impact‐R [Cone and Plate(let) Analyzer]. Journal of Thrombosis and Haemostasis, 7, 1990–1996.
• Seligsohn, U., Zitman, D., Many, A. & Klibansky, C. (1976) Coexistence of factor XI (plasma thromboplastin antecedent) deficiency and Gaucher’s disease. Israel Journal of Medical Sciences, 12, 1448–1452.
• Shenkman, B., Savion, N., Dardik, R., Tamarin, I. & Varon, D. (2000) Testing of platelet deposition on polystyrene surface under flow conditions by the cone and plate(let) analyzer: role of platelet activation, fibrinogen and von Willebrand factor. Thrombosis Research, 99, 353–361.
• Varon, D., Dardik, R., Shenkman, B., Kotev‐Emeth, S., Farzame, N., Tamarin, I. & Savion, N. (1997) A new method for quantitative analysis of whole blood platelet interaction with extracellular matrix under flow conditions. Thrombosis Research, 85, 283–294.
• Wright, S. (1992) Adjusted P‐values for simultaneous inference. Biometrics, 48, 1005–1013.
• Yatziv, S., White, M. & Eldor, A. (1974) Lysosomal enzyme activities in platelets of normal individuals and of patients with Gaucher’s disease. Thrombosis et diathesis haemorrhagica, 32, 665–669.
• Zimran, A., Sorge, J., Gross, E., Kubitz, M., West, C. & Beutler, E. (1989) Prediction of severity of Gaucher’s disease by identification of mutations at DNA level. Lancet, 2, 349–352.
• Zimran, A., Altarescu, G., Rudensky, B., Abrahamov, A. & Elstein, D. (2005) Survey of hematological aspects of Gaucher disease. Hematology, 10, 151–156.
British Journal of Haematology 2011; 153(3): 372-8

|










/https%3A%2F%2Fassets.over-blog.com%2Ft%2Fcedistic%2Fcamera.png)