

Small Molecule Inhibition for Gaucher Disease: Emerging Role of Substrate Reduction Therapy for Lysosomal Storage Disorders

by Edwin H. Kolodny, MD
Introduction: Substrate Reduction Therapy
The Role of Small Molecule Inhibition in the Balance for Inborn Errors of Metabolism
by Marc C. Patterson, MD, FRACP, FAAN
Epidemiology of Lysosomal Storage Diseases
Lysosomal Storage Diseases: General Principles, Mechanisms of Disease
Lysosomal Storage Disease Mechanisms and Effects
Treatment Strategies for Liposomal Storage Diseases
Treatment Strategies for Liposomal Storage Diseases: Miglustat
Miglustat in Gaucher and Tay-Sachs
Effects of Miglustat
Clinical Trials and Long-term Experience With SRTs for Gaucher Disease
by Gregory M. Pastores, MD
Gaucher Disease: Management Issues, Therapeutic Approaches, and Treatment Goals
Gaucher Disease: Management Issues, Therapeutic Approaches, and Treatment Goals (cont'd)
From the Literature: Studies of Substrate Reduction Therapy
From the Literature: Studies of Miglustat
Miglustat as an Active-Site-Specific Chaperone
Conceptual Approaches for the Current Management of Gaucher Disease

Slide 1. Clinical Trials and Long-term Experience With SRTs for Gaucher Disease
I want to focus primarily on Gaucher disease and in this presentation think of Gaucher disease as the paradigm for how we have managed the care of our patients in the short and the long term. I was among the principal investigators for recombinant glucocerebrosidase, which is the current standard of care for Gaucher disease.

Slide 2. Gaucher Disease
The diagnosis of the lysosomal storage diseases essentially relies on pattern recognition, recognition of the nature of the substrates that accumulate, and the sites of the accumulation or storage deposits. And, in fact, with Gaucher what you have is a continuum or a broad spectrum of clinical presentations that represent the outcome of storage and activation of cells derived from monocyte and macrophage lineage, primarily hematopoietic elements.

Slide 3. Gaucher Disease: Management Issues
But with respect to management issues, I think we all recognize that Gaucher disease is clearly a chronic disorder, particularly in the type 1 non-neuronopathic subtype, but is confounded by wide variability in clinical presentation and disease progression.
We know that the primary insult to cells is a consequence of glucosylceramide accumulation. But there is a cascade of downstream events that probably ultimately result in a crossing of a threshold that leads to the type of complications, such as avascular necrosis and interstitial lung disease or even primary pulmonary hypertension and perhaps parkinsonism as well, that we have encountered in our daily practice.
But to dissect these elements we really have to have a better understanding of what the drivers of morbidity are and, in practice, careful means of assessment to define, in essence, the pattern and severity of disease involvement. Obviously, understanding that risks may be different for early-onset patients, again, partly influenced by the nature of their underlying mutation.
And, obviously, when one decides to recommend treatment, I think it is quite important beforehand to define your treatment goals.
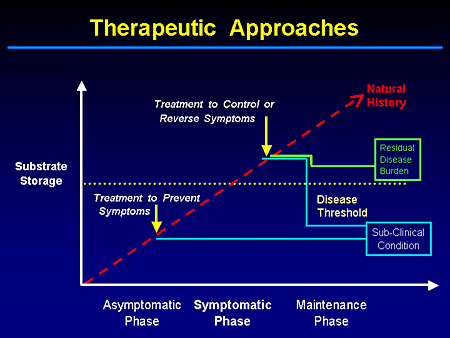
Slide 4. Therapeutic Approaches
This is a cartoon that many of you have seen before. And the goal ideally would be to prevent reaching this threshold or precipice by early intervention. And if we do have, and I guess we do, treatments that are relatively safe and effective, we may enable a subclinical threshold. We have not corrected the underlying gene or biochemical defect, but we prevent the onset of disease-related complications.
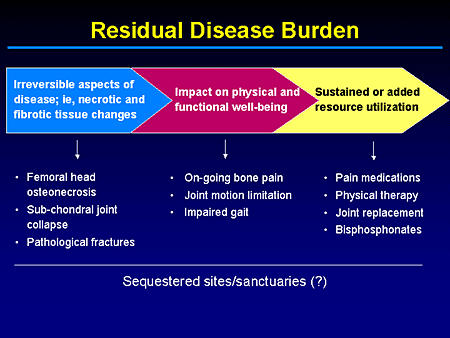
Slide 5. Residual Disease Burden
Alternatively, what we may end up with, in treatments that are partially effective or started too late, is this residual burden of disease. And if you will indulge me, I want to underscore this point again. We are not here to talk about pharmacoeconomics, but when you talk about overall comprehensive treatment plans, it's not something you can readily ignore with the type of agents that are out there. And in this realm when you talk about quality-of-life adjusted years or health-adjusted years, you kind of have to discount for things that may be irreversible that are the consequence of end-stage disease processes such as necrotic and fibrotic tissue changes.
I think fundamentally most treaters miss the fact that enzyme- and even substrate-reduction therapy relies on means to enhance clearance or reduce accumulation of storage material. And you still need the body's intrinsic ability to repair itself to affect ultimately some control, if not reversal, of the symptomatology.
But there may be challenges, such as dealing with the brain and the lungs, for example, and even bone. And whether this represents sequestered sites that the drug is not fully able to access or sanctuaries that are not as responsive to your therapeutic modality of choice is hopefully something we will clarify down the line.

Slide 6. General Management Goals
This is an exercise that I have to go through each time I recommend treatment. I think we all recognize this to be a genetic disorder for which we need to provide appropriate counseling. And unfortunately, we are dealing with a disease where the underlying mutations do not necessarily allow us absolute confidence in predicting outcome. But obviously, part of early intervention is recognizing potential family members at risk who may benefit from treatment.
And optimizing long-term outcome involves directed or adjunctive therapies, but we should not overlook the role that certain therapies, such as bisphosphonates, may have in addressing other manifestations, such as osteopenia, that may lead to increased risk for pathologic fracture in patients with Gaucher disease.
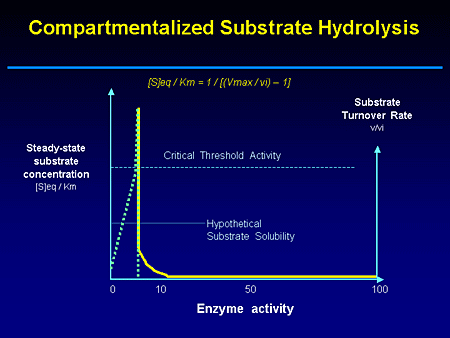
Slide 7. Compartmentalized Substrate Hydrolysis
This is a cartoon of this threshold being reached when a critical amount of storage material has, in essence, triggered those cascade events that I alluded to earlier. In fact, one readily appreciates by looking at this cartoon how you can have carriers who may on average have only 50% residual enzyme activity, but not show clinical manifestations for this autosomal recessive disease, and others with mild or deleterious mutation having different rates of disease progression.
It also becomes evident that there are at least 2 ways of treating this disease; by either supplementing enzyme activity or controlling substrate flux in and out of the lysosomal compartment. But I'll give you one of my quotes. "We are in an era in the treatment of lysosomal storage disease where we have current and emerging treatments." And as my niece tells me, "Enzyme therapy, it's so 1990s." But as I alluded to earlier, it remains the standard of care. But with enzyme replacement therapy the window of opportunity is opening not just for enzyme replacement, but perhaps for enzyme enhancement as well.

Slide 8. Recent Publications on SRT and GD
I wanted to call attention to at least 4 papers, highlights of which I will go through in my presentation, that reflect recent literature on substrate-reduction therapy and Gaucher disease. This does not necessarily represent the order of importance. I will, obviously, talk primarily about my experience.

Slide 9. Clinical Therapeutics
But what I'd like to do is highlight some of the points from this 24-month clinical trial of patients, either naive or patients whose enzyme-replacement therapy had been discontinued for one reason or another because of immunoglobulin E (IgE) formation in at least 2 patients.
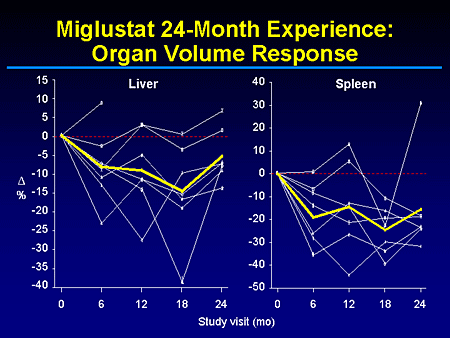
Slide 10. Miglustat 24-Month Experience: Organ Volume Response
This slide shows you a summary of the organ volume response, clearly showing a reduction in liver and spleen volume. And, in fact, most of the individual lines parallel the changes we see in liver and spleen with this patient who sort of reverted in both liver and spleen during the last 6 months of the trial, essentially representing an individual who has failed to comply with the rigors of therapy, which in this trial was 100 mg of miglustat 3 times a day.
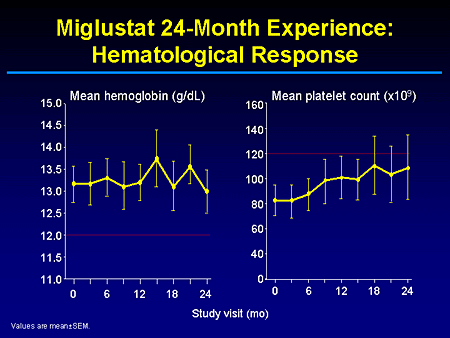
Slide 11. Miglustat 24-Month Experience: Hematological Response
And as you can see, these were patients who relatively had mild-to-normal hemoglobin measures that were effectively maintained during the trials and mild-to-moderate thrombocytopenia that was also stabilized or improved.
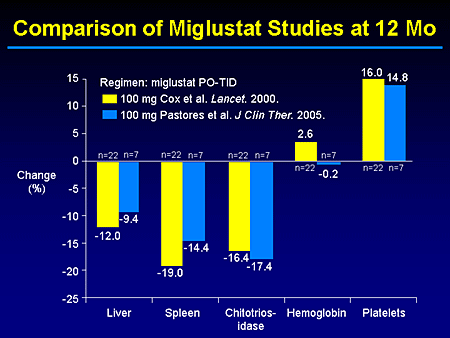
Slide 12. Comparison of Miglustat Studies at 12 Months
In essence, our trial was to demonstrate or confirm the earlier findings from the European experience by Cox and at other sites in Europe that also have been published.
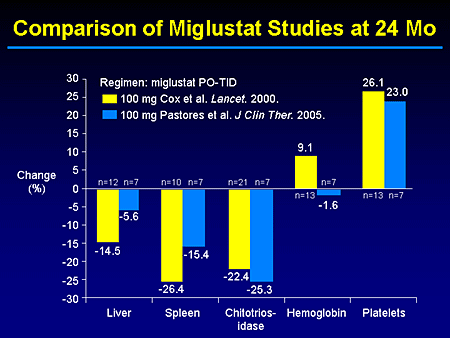
Slide 13. Comparison of Miglustat Studies at 24 Months
I think one will agree that at both the 12- and 24-month time point, our local, more limited experience with 7 or 8 patients monitored for up to 2 years essentially mirrors what has been reported by our European colleagues.

Slide 14. Sustained Therapeutic Effects of Oral Miglustat in Type I Gaucher Disease
In fact, Elstein from Shaare Zedek Medical Centre, Jerusalem, Israel, with Zimran and the other collaborators from the initial trial have recently reported the extended experience beyond 24 months, and have demonstrated a continued, persistent control of the disease with either stabilization or continued reversal of the clinical manifestations and effectively with no new safety concerns.
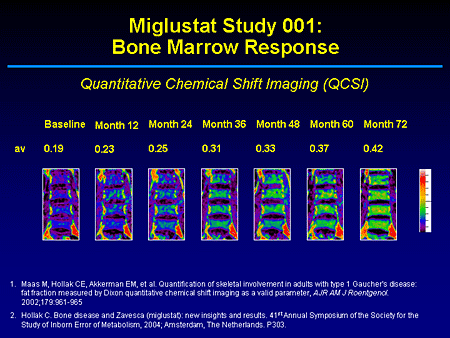
Slide 15. Miglustat Study 001: Bone Marrow Response
This paper is particularly interesting because for at least 2 patients we see for the first time within a short period of observation, in this case over 72 months, a reduction in the marrow burden of disease. What this shows are sequential magnetic resonance images (MRI) of the lumbar spine with quantitative chemical shift imaging and the application of the modified Dixon method. This allows you to measure the degree of displacement of adipocytes, which are primarily or predominantly triglycerides by glucosylceramide.
In the untreated patient this is what you will predominantly see on T1. Instead of a higher signal representing fat or yellow marrow in the axial skeleton, what you see is a marrow that is primarily of high water content and infiltrated with glucosylceramide. Over time what you can see is essentially close to normal imaging changes telling us that this is a compartment that miglustat can access. Dr. Patterson had alluded to its wide volume of distribution and that it is efficacious within the boney compartment where the storage material primarily is located.

Slide 16. Miglustat Experience: Safety Considerations
With regards to safety, I know when the trial was first reported, it got a lot of bad press, but now we appreciate that most of those concerns really were representative of the failure to include elements in the baseline assessments of patients that would have addressed potential safety concerns. But for those of you who do clinical trials and care for these patients, there has to be ongoing recognition that no matter how great our experience is, this is not hypertension or diabetes and we will need to remain vigilant. With respect to this product, there have been in our trial no clinically relevant abnormalities on serial neurologic examination, not just physical examinations by a board-certified neurologist, but electrophysiologic testing as well. And, in fact, in the Elstein report there were no new cases of peripheral neuropathy.
In Europe, there is a postregulatory commitment to monitor for any safety concerns through an intensive surveillance system, commonly known as the IS3, and there really has been no signal to indicate new concerns or reasons to withdraw the product.
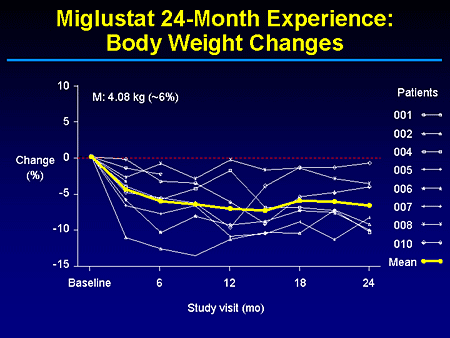
Slide 17. Miglustat 24-Month Experience: Body Weight Changes
I know you heard of some weight loss and it's not clear to us whether this is necessarily due to the diarrhea because that's usually during the first 10 days or 2 weeks of drug exposure. And, in fact, with dietary changes and timing of when people take these medications most of that is addressed. But as you can see here, this is not a persistent weight loss. There is some stabilization and I guess most of us can, in fact, probably benefit from 5% reduction in our current body weights.

Slide 18. Active-Site-Specific Chaperone (ASSC) Therapy: Enzyme Enhancement Therapy (EET)
I want to end with the final message that when the trials with the use of miglustat were first conducted, the proof of principle relied on the preclinical experience in mouse models of GM2 storage, whether due to a primary enzyme deficiency or secondary accumulation from a transport defect.
But as most of you may be aware in the Alfonso and colleagues paper from the study group in Zaragoza, Spain, they recently reported that miglustat, in fact, besides being an inhibitor of substrate synthesis by acting on ceramide-specific glucosyltransferase also may function as an active-site-specific chaperone.
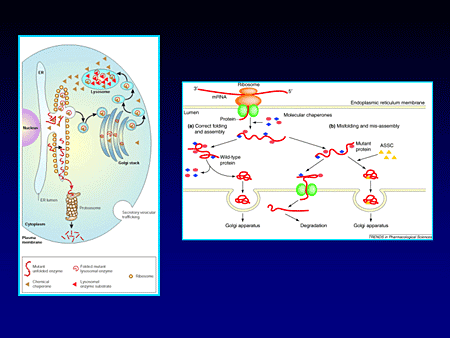
Slide 19.
The principle behind this essentially is the fact that there are certain mutations that are inactive in the lysosome because during their synthesis and ultimate need for transport to that acidified environment within the lysosome they are misfolded, diverted to the pathways of degradation, and perhaps, in fact, might trigger other cascades of other pathomechanisms by activation of the endoplasmic reticulum stress response.
But it turns out that miglustat is able to occupy the active site, enabling the appropriate refolding of certain mutant glucocerebrosidase, enabling its delivery to the lysosome where it remains functional and, in fact, these are transfected across cells.
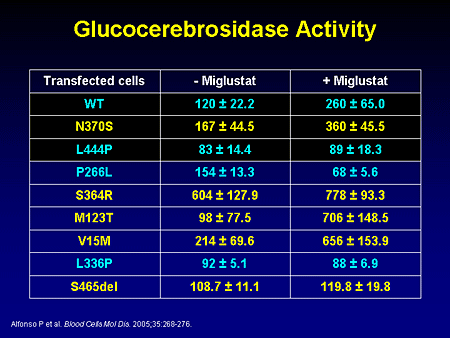
Slide 20. Glucocerebrosidase Activity
As you can see, there is enhancement not only of certain mutant alleles, but of wild-type alleles as well. Perhaps there are certain typographical errors even in a system that may have high fidelity that is somewhat corrected by this small molecule. But as you can see, it does not work uniformly and, thus, may not be a sole mechanism of action sufficient to control/reverse disease.
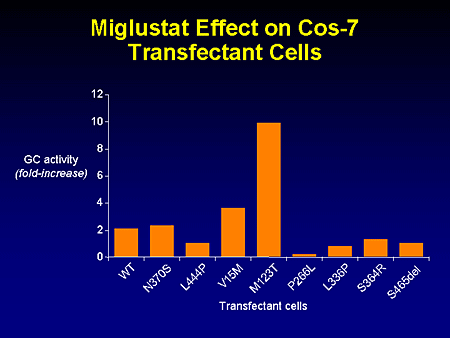
Slide 21. Miglustat Effect on Cos-7 Transfectant Cells
This just shows the full increase in glucocerebrosidase activity for the mutants that were examined, including not only the common, but rare alleles as well.
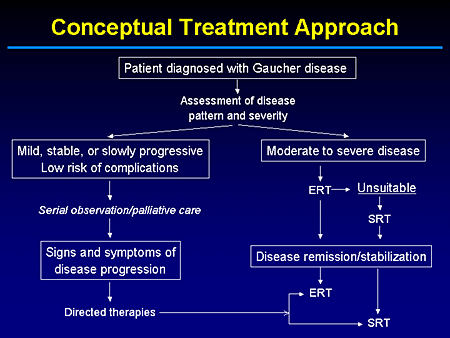
Slide 22. Conceptual Treatment Approach
This is a scheme in terms of conceptual approaches for the current management of patients with Gaucher disease.
The question obviously is, when you have a patient who has been on enzyme therapy from the beginning, meaning its commercial approval, do we feel our patients should continue to be committed to intravenous replacement therapy? Perhaps for most cases that should be the case, but this is an issue I think that we almost have a moral imperative to address.
In fact, I cannot go into great detail now because we are awaiting Institutional Review Board (IRB) approval, but there will be a multicenter study to, in essence, look at whether patients who have been stabilized by enzyme replacement therapy can, under a clinical trial protocol over a 2-year period, be effectively maintained by substrate-reduction therapy or miglustat monotherapy.
The concept that underlies this is the fact that when you treat a patient who is naive to either enzyme or an iminosugar, what you really are trying to deal with is preexisting material and preventing reaccumulation.

Slide 23. Conceptual Approach
But if enzyme-replacement therapy has done the most that it can in terms of clearing preexisting storage material, we need to know what the dose is, the frequency of infusion, or alternative therapies if what we are dealing with in the maintenance phase is just preventing reaccumulation.
I am not advocating a standard of care. I am just sharing with you what to me represent, not the end of science of Gaucher disease, but knowing what remains to be discovered.
Emerging Avenues for SRT in Inherited LSDs
by Cynthia J. Tifft, MD, PhD
Substrate Reduction Therapy in Lysosomal Storage Diseases
The Gangliosidoses
Phenotypic Spectrum of the Gangliosidoses
What We've Learned From Mouse Models
Mice Treated With Miglustat
Miglustat in Patients With Infantile Onset GM2 Gangliosidosis
Case Study: J.W.
Paradigms for Substrate Reduction Therapy in Lysosomal Storage Diseases

Slide 1. Emerging Avenues for SRT in Inherited LSDs

Slide 2. Lysosomal Storage Disorders
You can pick out your own favorite lysosomal disease from among this list. This is my way of going over the approximately 1 in 7700 individuals affected with lysosomal diseases. Notice that the vast majority of these have central nervous system (CNS) involvement and indeed for those that are not starred (for example, Gaucher disease type 1), one might wonder if they also have a CNS phenotype. So I would say the vast majority of these disorders do involve the CNS at some level.

Slide 3. Approaches to Therapy for LSDs Affecting the CNS
Dr. Kolodny and my co-speakers already have gone through most of these.

Slide 4. Emerging Avenues for SRT in Inherited Lysosomal Storage Disorders
What I'd like to do is review the biochemical basis and clinical phenotype of GM1 and GM2 gangliosidosis, which I'm interested in and which are probably the most difficult or some of the most difficult lysosomal diseases to think about treating; describe a genetic "proof of concept" for substrate-reduction therapy (SRT) that was conducted in the Proia laboratory several years ago; summarize the results of SRT in mouse models of both GM1 and GM2 gangliosidosis; discuss an ongoing clinical trial with miglustat in infantile patients with GM2 gangliosidosis that we have recently begun; and lastly, present a case of a child who I have known about probably for the last 5 years who has juvenile GM1 gangliosidosis and who has probably been on miglustat longer than any other non-Gaucher patient.
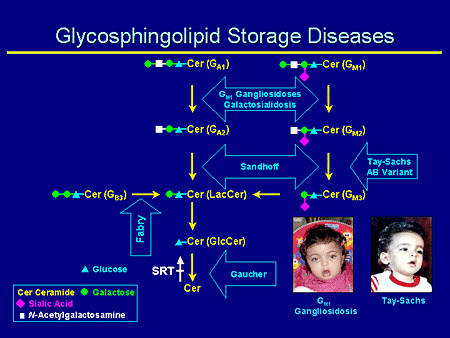
Slide 5. Glycosphingolipid Storage Diseases
Glycosphingolipid (GSL) storage diseases; this is my version of the complex gangliosides. The diseases that we're particularly interested in here are GM1, Sandhoff, and Tay-Sachs disease. Remember that SRT acts here at the first step and commits a ganglioside synthesis.
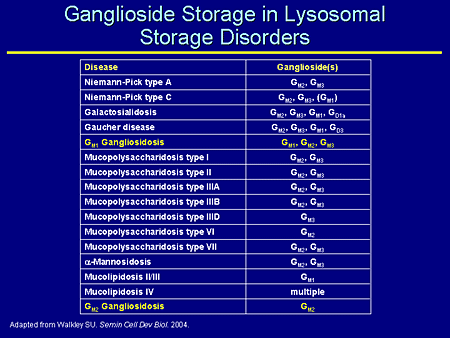
Slide 6. Ganglioside Storage in Lysosomal Storage Disorders
This is a list of ganglioside storage in a number of lysosomal storage diseases. This was adapted from a paper by Walkley where he nicely shows not only the primary gangliosidosis in which we're interested, but a number of lysosomal storage diseases that are not considered primary ganglioside storage diseases, which secondarily also store complex gangliosides.
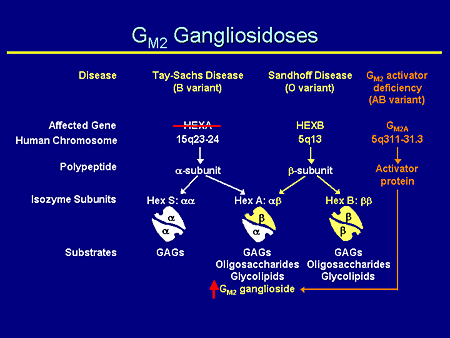
Slide 7. GM2 Gangliosidoses
The GM2 gangliosidoses are complicated; the nomenclature probably is more complicated than anything else. There are 3 specific diseases: Tay-Sachs, Sandhoff, and the much more rare GM2 activator deficiency. Each of them has an affected gene on a separate chromosome and chromosomal location. Each produces a separate gene product: the alpha, the beta subunits, and the activator protein. And each of them forms together in combination: hexosaminidase S, the alpha/alpha dimer; hexosaminidase A, the alpha/beta heterodimer, which is, incidentally, the only enzyme that can degrade GM2 ganglioside; and lastly the beta/beta homodimer, which can degrade glycosaminoglycans, oligosaccharides, and glycolipids.
Therefore, if one has a mutation in the hexosaminidase A gene, one has a decrease in the alpha subunit and an increase in the amount of GM2 ganglioside.
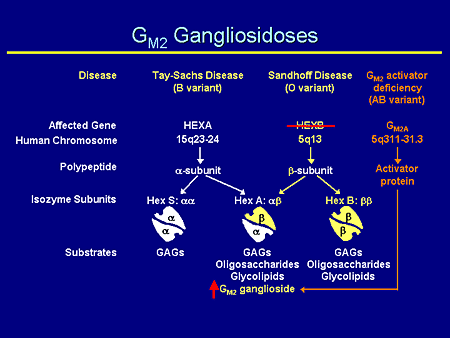
Slide 8. GM2 Gangliosidoses
Likewise, if one has a mutation in the hexosaminidase B gene, one sees a decrease in the amount of the beta subunit and again, an increase of GM2 ganglioside because it is only hexosaminidase A that is able to degrade GM2 ganglioside. But you also will notice here, and this will become important later, that the beta/beta homodimer also is decreased, and that's responsible for the degradation of glycosaminoglycans and oligosaccharides.
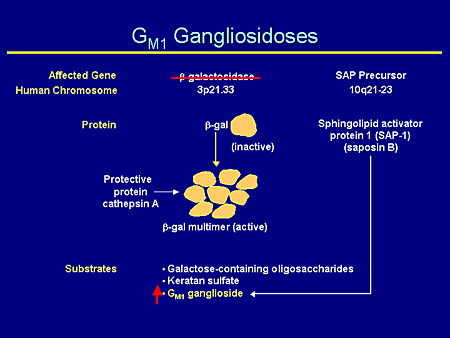
Slide 9. GM1 Gangliosidoses
GM1 gangliosidosis, likewise, has a unique effective gene product, beta galactosidase, on the short arm of chromosome 3. It produces an inactive subunit; when together in a multimer with protective protein cathepsin A, it produces an active enzyme and degrades galactose-containing oligosaccharides, keratan sulfate, and GM2 ganglioside such that mutations in the gene produce a decrease in the amount of active subunit and an increase in GM1 ganglioside.

Slide 10. Phenotypic Spectrum GM2 Gangliosidosis
As has been mentioned many times for all of these lysosomal diseases, both GM1 and GM2 gangliosidosis reflect a phenotypic spectrum, which is related to the residual enzyme activity. The less enzyme activity you have, the worse your disease. And this is, of course, on each person's unique genetic background which, again, provides even more variability in the disease.
The classic infantile form has an onset at less than 6 months; a startle response; hypotonia; a cherry-red macula, which often is the way these children come to diagnosis from the ophthalmologist; seizures that begin usually after 1 year of age; macrocephaly beginning about 18 months of age; progressive visual loss; and death between 2 and 5 years of age, depending on the kind of supportive care the family chooses to provide.
Juvenile is more variable: onset 1.5 to 5 years of age, cerebellar ataxia, cognition impairment, visual loss, seizures, and death in the mid teens.
Even more variable are adults with chronic onset form, which is onset, generally speaking, in early adulthood; progressive weakness; cerebellar ataxia; and psychosis, which is important because often these patients present with psychosis, and this is a problem for several years before the diagnosis actually is made. The age of death is quite variable.

Slide 11. Phenotypic Spectrum GM1 Gangliosidosis
Likewise for GM1, again, there is a spectrum of disease severity. The GM1 infantile patients actually are more severe than the GM2 infantile patients, with onset at less than 6 months. They are the children who can be born with dysmorphic features, skeletal dysplasia, hepatosplenomegaly; their developmental arrest is probably a little earlier; hypotonia progressing to spasticity; again, a cherry-red macula, which is how they come to diagnosis; seizures; and death usually at less than 2 years of age.
There is more variability in the late infantile/juvenile form, as you can see here, similar to the juvenile form for GM2.
And the chronic or adult-onset form is even more variable yet with onset between 3 and 30 years of age, with some skeletal changes, intellectual deterioration, and variable ages at death.

Slide 12. Phenotypes of GM2 Knockout Mice
Now we're going to switch to the mouse models and what has been known about the mouse models. This work has been done probably over the last 5 to 10 years, some of it in the Proia laboratory, some of it in other laboratories. But early on there were 3 different types of knockout mouse: the knockout mouse for Tay-Sachs, which was a deficiency of the XA gene; a knockout mouse for Sandhoff or XB null mutants; and the activator deficiency.
As it turns out, in the human situation, Sandhoff and Tay-Sachs are roughly equivalent with the exception that Sandhoff also features hepatosplenomegaly and oligosacchariduria.
In the mouse, the Sandhoff mouse is much more severely affected than the Tay-Sachs mouse. And as Dr. Patterson already has alluded to, the Tay-Sachs mouse doesn't have a clinical phenotype, but has a histopathologic phenotype that has been somewhat useful. The Sandhoff mouse, on the other hand, appears normal to about 2 months and then has an impaired rotor rod test, impaired motor function, develops ataxia, muscle weakness, hind-limb paralysis, head shaking, and dies at just prior to 4 months.

Slide 13. Genetic "Proof of Concept" for SRT
In describing the genetic proof of concept for substrate reduction, I go back to something that I use with my patients in trying to help them understand SRT. And excuse me if this is way too simplistic for this audience. But if the red circle is the amount of residual enzyme or the amount of enzyme you have if you make substrate, you have plenty of enzyme in order to degrade it. If you have disease, you have a deficiency in enzyme. If you have the usual amount of substrate that you can't degrade, you store. And if one uses SRT, one does not change one's amount of enzyme degradative capacity. One simply turns down the faucet and decreases the amount of synthesis.
So the genetic proof of concept was to breed a mouse that was not able to synthesize complex gangliosides with a mouse unable to degrade GM2 ganglioside, ie, the Sandhoff mouse. And in that way one could create a mouse that was genetically substrate deprived.

Slide 14. Glycosphingolipid Synthesis
This is an example. SRT blocks at the first committed step and synthesis. This mouse actually was made by making a null knockout for the N-acetylgalactosaminyltransferase I (GalNAcT) gene and basically this mouse produces no complex gangliosides.
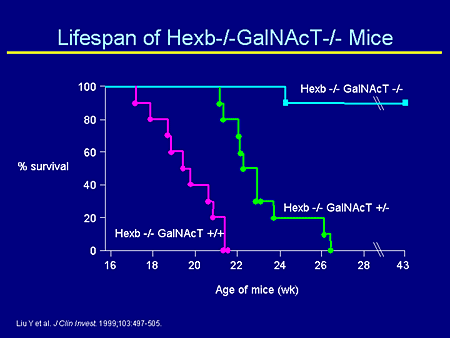
Slide 15. Lifespan of Hexb-/-GalNAcT-/- Mice
If you breed this mouse with a Sandhoff mouse, what you can see is the following. The untreated Sandhoff mouse dies at about 4 months. The mouse that was the double knockout has Sandhoff and is now double knocked out for the GalNAcT, and this mouse lives for a long time. And, in fact, what the mouse finally succumbs to is a storage of oligosaccharides, not a storage of gangliosides.

Slide 16. Double Knockout Mouse Lacks GM2 Storage
As you can see here in the gel, the wild type synthesizes in an array of complex gangliosides. The Hexosaminidase-B mouse stores GM2 ganglioside. The GalNAcT knockout does not make complex gangliosides, but makes increased amounts of GM3. And the double knockout mouse does not store GM2 ganglioside because it does not make GM2 ganglioside.
So the genetic proof of concept is not the same thing as SRT for 2 reasons. The genetic construct is a complete knockout and the genetic construct never synthesizes complex gangliosides. In the SRT model, there is no deficiency in your ability to make complex gangliosides, and this is not something that starts at point A; this is something that one adds later, usually when storage already has occurred.
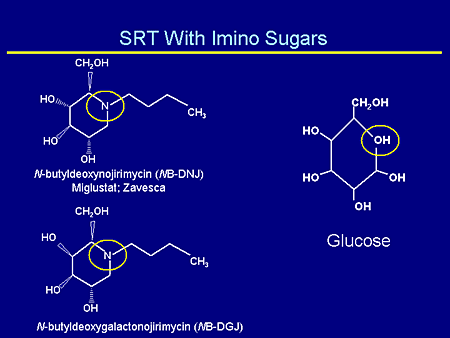
Slide 17. SRT With Imino Sugars
My colleagues already have alluded to the different types of imino sugars that have been used: N-butyldeoxynojirimycin (NB-DNJ) (miglustat or Zavesca), and N-butyldeoxygalactonojirimycin (NB-DGJ), the galactose derivative, both of which mimic glucose.
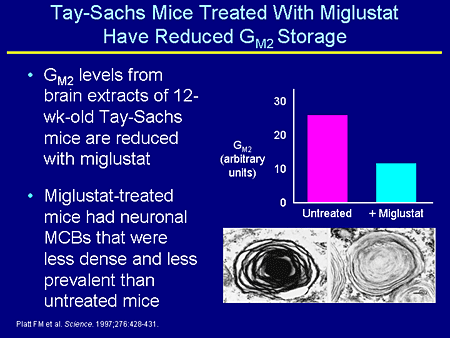
Slide 18. Tay-Sachs Mice Treated With Miglustat Have Reduced GM2 Storage
Initially this was work by Platt where they took the Tay-Sachs mouse, the one that does not have a clinical phenotype, and treated this mouse with miglustat. And you can see a decrease in the amount of GM2 and a decrease in the intensity of the multilamellar cytoplasmic bodies in treatment with miglustat in Tay-Sachs mice.
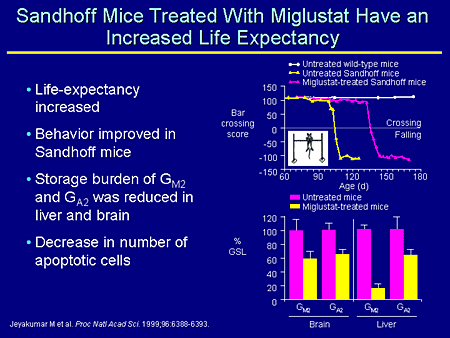
Slide 19. Sandhoff Mice Treated With Miglustat Have an Increased Life Expectancy
In the Sandhoff mice you see a similar thing, although we now have a phenotype that we can look at, and these mice have an increased life expectancy. The behavior, which is a bar crossing test, is improved in the Sandhoff mice treated with miglustat, and the storage burden is much reduced both in brain and in liver. Indeed, there is a decrease in the number of apoptotic cells in the brain.
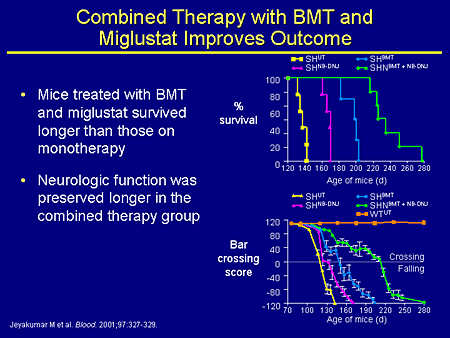
Slide 20. Combined Therapy with BMT and Miglustat Improves Outcome
We undertook a combined experiment with Platt's group several years ago where we performed a bone marrow transplant; you just about double the lifespan of the mouse by doing a bone-marrow transplant between 10 and 14 days of life. And if you take those transplanted mice and treat them with miglustat, you can get a synergistic effect both in survival and in behavior. So this sort of speaks to the idea of combined therapy. That might also be something we need to be looking at clinically.
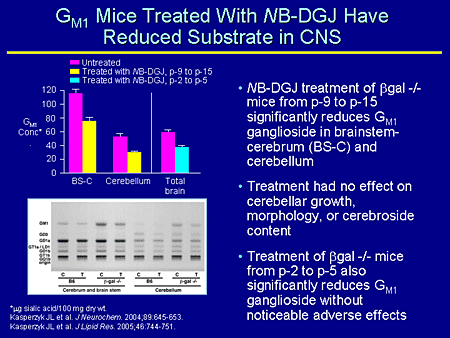
Slide 21. GM1 Mice Treated With NB-DGJ Have Reduced Substrate in CNS
Let's turn to GM1 mice; this is a knockout for GM1. These mice were treated with a galactose derivative and have reduced a substrate in the CNS. This was work that was reported recently by Seyfried's group at Boston College, Chestnut Hill, Massachusetts, and these were 2 separate experiments. One was treating with NB-DGJ; mice at postnatal day 9 (p-9) to p-15. So these are very short treatment periods and they were looking at brainstem cerebrum and cerebellum, both of which saw a considerable decrease in the amount of GM1 concentration.
In a second experiment they looked at in even younger mice, at p-3 to p-5, and again saw a decrease in total brain.
The gel is from the p-9 to p-15 and you can see the wild-type mice, but in the knockout mice you can see a reduction of GM1 in the mice treated with NB-DGJ.
Furthermore, Seyfried makes the point that treatment appeared to have no effect on cerebellar growth, morphology, or cerebroside content in either of the mice treated at either time. One would be hopeful that this might be an important therapeutic regimen for patients with GM1 gangliosidosis.

Slide 22. SRT With Imino Sugars in the Treatment of LSDs With CNS Involvement
To summarize, there are a number of mouse models that have been treated with SRT: GM2, Tay-Sachs and Sandhoff; GM1; and Niemann-Pick C. And clinical trials are ongoing in these disorders. The one I'm particularly interested in is the one that's going on at Children's National Medical Center, Washington, DC, which I am going to tell you about.

Slide 23. Pharmacokinetics, Safety, and Tolerability of Miglustat in Patients With Infantile Onset GM2 Gangliosidosis
This is called "Pharmacokinetics, Safety, and Tolerability of Miglustat in Patients With Infantile GM2 Gangliosidosis." Do not get me wrong. This is not going to cure infantile GM2 gangliosidosis and we know that, but we want to see how this drug is metabolized and how young children and infants are able to tolerate this drug.
The inclusion criteria are a confirmed diagnosis of GM2, either Tay-Sachs or Sandhoff disease, and onset of symptoms before age 9 months. So these would be infantile patients. They have to be within 1 year of diagnosis and less than 2 years of age. We don't want children who are very ill for their own safety. So we want to take children who are relatively healthy. The study lasts for 6 months and we want to make sure they'll be able to complete the study. They need to have normal renal and hepatic function and informed consent.
What will exclude these children is if they are receiving any other investigational agents; if they have a significant history of a gastrointestinal (GI) disorder or clinically significant diarrhea for obvious reasons (this is one of the major side effects); patients who are anemic because doing pharmacokinetics we're going to take a reasonable amount of blood from these children, not unsafe, but reasonable amounts (we don't want them to be anemic); and patients who have a high probability of dying during the 6-month assessment, which again is the reason for choosing earlier children.

Slide 24. Pharmacokinetics, Safety, and Tolerability of Miglustat in Patients With Infantile Onset GM2 Gangliosidosis
The outcome measures are miglustat levels in matched plasma and cerebrospinal fluid (CSF) samples at baseline and at steady state (this will be sort of the first systematic look at how much miglustat is able to get into the CSF in these children); GM2 ganglioside levels in plasma and CSF at baseline and at 6 months as determined by tandem mass spectrometry; assessment of biomarkers including inflammatory markers such as chitotriosidase that we know is increased in the CSF of patients with GM2 gangliosidosis; progression of the cherry-red macula using digital photography; neurologic assessments including electroencephalogram (EEG), visual and brainstem evoked responses; and, obviously, developmental evaluations.

Slide 25. Miglustat and GM1 Gangliosidosis
Now I'm going to tell you a story, and the story I think emphasizes 2 things. One, that it may be possible to use miglustat in the treatment of patients with GM1 gangliosidosis. And two, I think it speaks to the tenacity of our patients and their families in being able to search the Internet and find any possible therapy for their children, and I think the faith they place in scientists and doctors in being able to find cures for their children.
This is taken from a diary that a mother sent to me. This is a mother I've been communicating with for about the last 5 years. I've never met this child. I hope to very shortly. The mother's diary was corroborated by her developmental pediatrician, who is prescribing the miglustat, and the child's therapist.
J.W. was born to a 41-year-old mother following an uncomplicated pregnancy. Psychomotor development was normal or you might say advanced. He sat at 5 months, crawled at 6 months, and walked at 10 months. By 2 years of age he had a large vocabulary, could sing songs, and memorize Bible verses and poems. He was doing elementary school workbooks at age 4 years.

Slide 26. Miglustat and GM1 Gangliosidosis
At age 5 years, J.W. started in Little League, but by the end of the season began stumbling and he began to stutter. At entry into kindergarten he was able to write his name, but this ability was lost by the end of the year. He began to regress in all areas, including running. He began to walk flexed at the hips with a rather rounded back.

Slide 27. Miglustat and GM1 Gangliosidosis
Magnetic resonance imaging (MRI) at age 10 years showed cerebral atrophy. EEGs were abnormal. He was initially diagnosed with Landau-Kleffner syndrome, but at age 11 years an ophthalmologic evaluation revealed clouded corneas. Blood work was sent to the laboratory for lysosomal enzyme analysis, and the deficiency of beta galactosidase was reported, therefore confirming the diagnosis of juvenile GM1.

Slide 28. Miglustat and GM1 Gangliosidosis
From age 12 to 15 years, he continued to deteriorate. His speech regressed to 1-word responses, then to grunts, and then to words that hardly anyone could understand, even his mother. He went from regular classes to partial, then full, then a complete special education program, and finally to a classroom for profoundly impaired children. He lost the ability to feed himself.
His mother describes at about age 15 years he was really out of it. He would sit in his wheelchair all day or nap, and had lost toilet training. He couldn't communicate and just sort of sat there.

Slide 29. Miglustat and GM1 Gangliosidosis
At age 16 years, J.W. began taking miglustat at the recommended dose for Gaucher disease, 100 mg 3 times daily. January 2004 was the same month that miglustat was US Food and Drug Administration (FDA)-approved for the treatment of Gaucher disease. His mother was really on top of this.
Within the first month his energy level and alertness increased, and in that same month he began to walk. One day he just stood up and moved from the bedroom to the living room; this child had not walked in 4 months.
By month 2, mother reports that the therapist indicated he had regained some computer skills. His eyesight had improved and he was regaining language, initially single words and then phrases. And if you remember, he had lost his language abilities.

Slide 30. Miglustat and GM1 Gangliosidosis
Over the past year J.W. has had enough increase in energy that he was able to participate in the Special Olympics and, indeed, this is first place in the wheelchair race. He is now almost completely re-toilet-trained after having completely lost the ability. He is now working at a large pet store and he has been hired by a local pet resort to do playtime with the dogs. And, as his mother remarks, he continues to gain skills.
I will tell you that he has continued to have more difficulty with his hips, which I think raises an important consideration in that, obviously, if we are impacting his storage of GM1, this is being impacted in his CNS. But probably the other things he is storing, such as dermatan sulfate, are contributing to his skeletal problems and that is not going to get better with miglustat.

Slide 31. Paradigms for SRT in Lysosomal Storage Disorders Affecting the CNS
Paradigms for SRT in lysosomal storage disorders -- this is a summary. SRT is more likely to be successful in individuals who have a greater amount of residual enzyme activity, ie, the adult-onset form. I think that's pretty clear. The hole in the can is a little bit bigger.
In individuals with little or no residual enzyme activity, SRT is likely to be successful only in combination with other therapeutic modalities, such as enzyme-replacement therapy (ERT), stem-cell transplantation, or molecular chaperones.
SRT instituted at the earliest possible time will maximize the chances of successful outcome. And here is my plug for newborn screening for lysosomal diseases, not just the ones we know are treated right now, but the ones who are going to be treated in the next 5 years.

Slide 32. Paradigms for SRT in Lysosomal Storage Disorders Affecting the CNS
In some GSL storage disorders, additional non-GSL products also accumulate. We just talked about that in the case of GM1. That's also true for GM2. Non-GSL product accumulation will not be affected by SRT and, indeed, where GSLs are the primary storage product and the primary pathology, SRT may unmask additional symptomatology as they have in this child, as they did in the double-knockout, GalNAcT Sandoff mouse that then died of the oligosacchariduria.
Lastly, GSLs also accumulate in lysosomal diseases that are not primary GSL storage disorders, as detailed in the chart by Walkley. And SRT may have a role in the treatment of the CNS component of these disorders as well, and here would be some of the mucopolysaccharidosis. Obviously, we're not going to completely reverse these, but it may help address the issue of ganglioside storage.
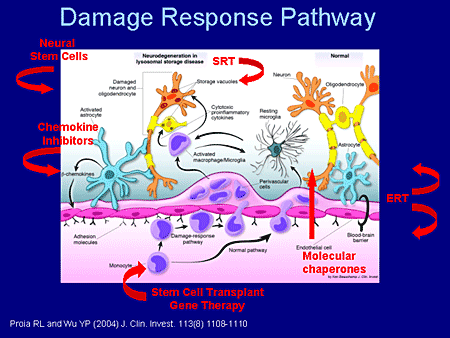
Slide 33. Damage Response Pathway
This represents work done in the Proia laboratory and was published in 2004. Let me walk you through it.
In the normal situation, in the normal pathway, monocytes go from the blood across the blood-brain barrier, which is very tight, and take up residence as resting microglial cells and participate in immune surveillance in the brain. These are normal neurons, normal oligodendrocytes, normal astrocytes.
In a situation like primary gangliosidosis, GM1 or GM2 storage produces a sick neuron (there are a lot of "black boxes" still in this diagram) that signal the astrocytes, and we know that. And the astrocytes now are activated to produce beta chemokines, which then traverse back across the blood-brain barrier into the circulation and actually recruit additional monocytes in this damage-response pathway. The macrophages are then activated. They themselves also are enzyme incompetent. They try to do their job and can't do it, and one then ramps up a huge inflammatory response.
So how might one try to intervene in this pathology? As I reflected before, this is going to include, more than likely, combined therapies. SRT would act at the level of prevention or ramping down the amount of ganglioside that's synthesized. Molecular chaperones, themselves small molecules, could travel from the vascular space into the CNS space and enhance enzyme activity. One also might envision stem cell transplants and gene therapy or engineered monocytes that would then superproduce enzymes that could be injected into the circulation and make their way across the blood-brain barrier, and now these would be enzyme-competent cells that would sort of ramp down the inflammatory response. We know this happens in the GM2 mouse.
To the extent of which it happens in humans, I can tell you that children who are transplanted presymptomatically actually have a very flat or no rise in chitotriosidase, an inflammatory marker, whereas children who are untreated have 50-fold elevations. So there is some hope in that way.
Neuronal stem cells, which would go directly into the CNS and which would be enzyme competent, would be another possible mechanism. Chemokine inhibitors that would ramp down the recruitment of enzyme-incompetent monocytes to decrease the inflammatory response also might be possible.
And lastly, I think we should not forget about ERT in this CNS context. There has been some work with direct injection of ERT in the mucopolysaccharidosis type I (MPS I) system into the CNS that's shown some promise recently.
And in a recent paper with the MPS-VII mouse, using larger doses of ERT than normal directly into the circulation actually has boosted the level of enzyme in the CNS and there has been some benefit seen from that as well.
So I think the take-home message here from what all of us have been saying is that there are a number of ways to approach the problem. Here are a number of ways that these might act. It's really going to be sort of disease specific with regard to which ones we pick and choose in terms of ultimate therapy.
Slide 2. Overview

This brief talk will begin with some discussion of the epidemiology of lysosomal storage diseases. I'll say a little about the general principles involved, something about the range of phenotypes and their correlation with the underlying biochemistry, mechanisms of disease, and then, of course, treatment strategies
Slide 3. Incidence of IEMs in BC: 1969-1996

I'm sure everybody is very well aware of this paper from British Columbia. This looked at the incidence of inborn errors of metabolism (IEMs) in that geographic region between 1969 and 1996 (Applegarth and colleagues). And essentially they found a rate of small molecule diseases of 24 per 100,000 followed by the lysosomal storage diseases, about 8 per 100,000 which, as you can see, almost exceeded the combined incidence of these other types of disorders: peroxisomal disorders, oxidative phosphorylation defects, and glycogen-storage diseases.
Of course, there also is a caveat because, as we all know, many of these diseases are not diagnosed until late and, in fact, the diagnoses may often be missed altogether, particularly in the adult or more subtle phenotypes. So I think most of us would agree that these are likely to be underestimates of the incidence of these diseases.
Slide 4. Prevalence of LSDs (Australia 1980-1996)

This study covered the prevalence of lysosomal-storage diseases, the work of Hopwood and colleagues in Australia, from January 1, 1980, through December 31, 1996, a 17-year period. During that time, they diagnosed 27 different lysosomal-storage diseases in 545 individuals. I should point out to you that during this time the mean population of Australia was around 17 million. It didn't pass the 20 million mark until 2000.
The prevalence varied, as is universally the case. Gaucher disease was the most common of the lysosomal storage diseases in this population, 1 in 57,000. Sialidosis is not a disease you'd want to be waiting for in the clinic, 1 in 4.2 million.
But the point I want to make is that although there is considerable variability in the prevalence of the individual diseases, if you combine the prevalence of lysosomal-storage disease, it comes out to 13 per 100,000 or about 1 in 7,000. These are not rare diseases collectively. And again, I would emphasize the fact that in many cases, particularly in the more subtle phenotypes, the diagnosis often is not made, and I suspect this is an underestimate. So devising an effective therapy for these diseases is a significant public health problem.
Slide 5. General Principles
Lysosomal Storage Diseases: General Principles, Mechanisms of Disease

What general principles can we invoke? First of all, I think that experience over the years in general would support the contention that the phenotypic severity is proportional to the extent of residual enzyme activity, or where the primary defect is not on an enzyme to the extent of residual function of the gene product. And that in turn is proportional to the extent of substrate accumulation.
It follows from that simple principle that an early-onset disease will be one in which there is very low or absent activity of the gene product, which is accompanied by extensive substrate accumulation and produces a phenotype with severe manifestations, early onset, and a rapid course.
Later-onset disease tends to correlate with a greater degree of residual activity, a lesser extent of substrate storage, and a more subtle, slowly progressive course. And these, in fact, are from the more difficult phenotypes to diagnose.
It also has been observed that relatively small increases in enzyme activity or the activity of the gene product, reduction of the substrate load, or both would be expected to produce a substantial improvement in the phenotype.
And a final point to bear in mind is that although these are general principles that apply to this family of diseases, the precise patterns of involvement are disease specific, which, of course, is relevant to making a clinical diagnosis.
Slide 6. Varying Thresholds for Specific Symptoms
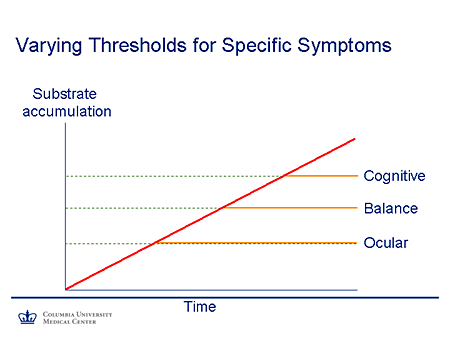
This is a simple graph that I find helpful in thinking about these diseases. We think of the body as a group of systems functioning in parallel, and the nervous system itself can be thought of as a family of systems that each function in parallel. Each system has its own intrinsic reserve capacity. And so we would expect that if a substrate accumulates and the reserve capacity is exceeded, symptoms will appear.
So, for example, in this case if we think of time on the x-axis (the extent of substrate accumulation on the y-axis as time passes), the most sensitive system -- in this case with some of the lysosomal-storage diseases, the eye-movement control system, which has relatively few neurons involved -- will be the first to manifest. As a substrate burden increases further, ataxia and other manifestations of cerebellar disease related to Purkinje cell dysfunction will become apparent. And finally, as the larger reserve pool of the cortical neurons becomes involved, cognitive impairment becomes apparent.
The slope of the line reflects the degree of residual activity of the gene product. So again, it follows that if there is relatively more residual activity, there'll be a slower course. But nevertheless, at least in theory, if the individuals survive long enough, symptoms would occur.
Slide 7. Classification of LSDs by Mechanism

If we think about mechanisms of lysosomal storage diseases, the classic mechanism described 40 years ago was that of an enzyme deficiency, specifically an acid hydrolase whose deficiency led to the accumulation of a variety of molecules. And it's the nature of molecules that led to the classification of diseases: proteins and the neuronal ceroid lipofuscinosis; glycoproteins and the glycoproteinoses, such as mannosidosis; the complex carbohydrates, the mucopolysaccharidoses; and lipids and the classic sphingolipidoses, such as Tay-Sachs, Gaucher, Niemann-Pick, and so on.
It may be that the enzyme activity is impaired and yet the gene coding for the enzyme is perfectly normal. In that case, the deficiency is in a cooperating protein, such as an activator protein, the saposins. This is the cause of the AB form of GM2 gangliosidosis, a rare phenotype. And more recently saposin A deficiency has been described for the first time as a cause of Krabbe disease.
There may be a problem in targeting the lysosomal enzymes to their final destination. This occurs where there is a problem with phosphotransferases and the Golgi apparatus so that multiple lysosomal enzymes are deficient in their site of action, although they then are directed along the secretory pathway and their concentrations are increased in the peripheral circulation; this is the mechanism of disease in the mucolipidoses.
Finally, there may be a quite different mechanism of action such that the endosomal lysosomal system, which is essential for transporting these large molecules in the otherwise unfriendly aqueous intracellular environment, is deranged. And we know now that Niemann-Pick disease type C is such a disorder, in which intracellular trafficking is abnormal. The precise mechanism is not yet understood, even in Chediak-Higashi syndrome, which typically presents with a hematologic/immunologic phenotype in the early onset disease, but a parkinsonian dystonia syndrome with later-onset disease.
Slide 8. Neurological Involvement in Gaucher Disease Type 1

So the principle I mentioned earlier, in terms of residual activity and the manifestations of disease, applies very nicely to Gaucher disease. We are familiar with the rare and very severe phenotype of type 2 Gaucher disease where enzyme activity is essentially absent. There's an antenatal onset of disease and death occurring within the first 2 years, an intermediate, if you will, neuronopathic form sometimes known as type 3 Gaucher disease. For a long time it was believed that type 1 Gaucher disease did not have a neurologic phenotype. But evidence has been accumulating recently, that there is a neurologic phenotype associated with type 1 Gaucher disease, but that it tends to appear in later life, consistent with that slow accumulation of substrate and a later onset of disease.
So sensorineural hearing loss used to be more common in these patients. Some have been reported with abnormal eye movements. Parkinsonism, which of course is common, does appear from early epidemiologic studies to be more frequent in patients with type 1 Gaucher disease. There is evidence from neuropathology of histologic lesions in the hippocampus. And so I emphasize the fact that the neurologic symptoms appear not to be restricted to types 2 or 3 and that, in fact, Gaucher disease, as you might expect, shows a phenotypic continuum, which really is the case for all of these diseases. The subtypes we use are helpful to us in thinking about these diseases, but they are a great oversimplification of the actual biology.
Slide 9. Secondary Mechanisms in LSDs

What secondary mechanisms might occur? Well, the substrate accumulation, particularly glycosphingolipids and the sphingolipidoses, has been correlated with inflammation in the central nervous system (CNS), something I think that has not been appreciated in the past; with abnormal calcium homeostasis, particularly calcium uptake; with structural abnormalities, which Walkley has so beautifully described in the form of ectopic dendritogenesis and meganeurite formation; and with accelerated apoptosis.
Slide 10. Inflammation and Impaired Calcium Homeostasis
Lysosomal Storage Disease Mechanisms and Effects
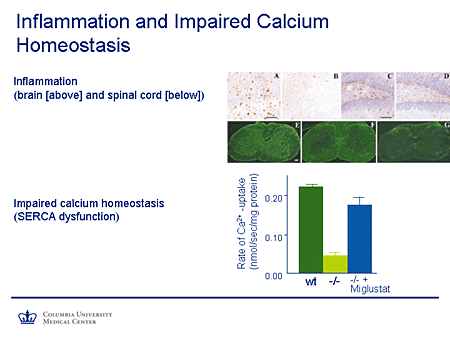
Here is some of the work of Platt and colleagues. Here in the section of cortex, the staining shows activation of markers of inflammation before and after treatment with substrate inhibition in this animal model using miglustat (N-butyldeoxynojirimycin [NB-DNJ]). In the hippocampus, again you can see the spots representing markers of inflammation. They are reduced, although not entirely absent, after treatment with miglustat; similar findings are seen in the spinal cord. These are all in animal models.
Similarly, I mentioned calcium homeostasis. The y-axis here represents the rate of calcium uptake in the system. In the wild-type animal you can see a robust level of calcium uptake. In the untreated mutant it's dramatically reduced, and there is significant recovery of function in the animal that is being treated with substrate-reduction therapy using miglustat
Slide 11. Ectopic Dendritogenesis
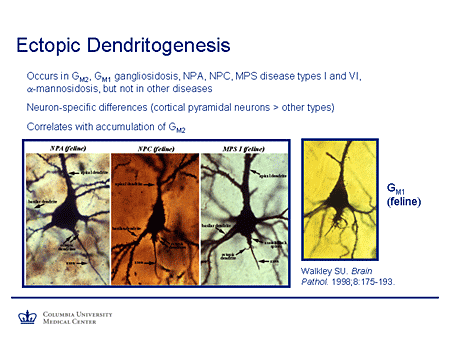
This is Walkley's work you see here, examples of ectopic dendritogenesis in giant cortical pyramidal neurons from feline models of lysosomal storage diseases; in Niemann-Pick A and C, mucopolysaccharidosis 1, and monosialoganglioside (GM1). And in all cases you can see these extra structures, which are ectopic dendrites, occurring in a location they would not normally be seen. And the interesting correlation demonstrated was that ectopic dendritogenesis occurs at sites where there is excess accumulation of GM2 ganglioside. Physiologically this is normally downregulated soon after birth when dendrite formation, at least to a large extent, is switched off. But in this case it's reactivated by the accumulation of this substrate. This type of neuropathology is not seen in lysosomal storage diseases in which GM2 ganglioside does not accumulate.
Slide 12. GM2 Accumulation, Apoptosis, and Microglial Activation in Sandhoff Mice
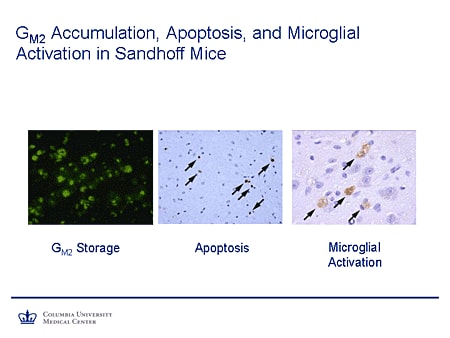
We also have mentioned apoptosis, microglial activation. You see here a stain from this section of brain. The green stain shows excess storage of GM2. The stains at the arrowheads (middle panel) show markers indicating active apoptosis, with the staining on the right showing evidence of microglial activation, all downstream consequences of excess substrate storage.
Slide 13. Conclusions

So we can conclude that the phenotype of the lysosomal storage diseases is proportional to the severity of the primary defect and, thus, proportional to the amount of excess stored substrate.
Secondly, the classic phenotypes are disease specific, and they will often allow an accurate clinical diagnosis requiring only specific confirmatory testing. It also seems to follow logically that other factors being equal, partial repair of the primary defect, reduction of storage substrate, or both would be expected to ameliorate the phenotype.
Slide 14. Treatment Strategies for LSDs
Dr. Kolodny already has alluded to the treatment strategies for lysosomal storage diseases. I won't belabor it too much, but will point out that definitive therapies might be those in the left-hand column, and supportive but nevertheless potentially effective therapies are in the right column. Of course, the "Holy Grail" would be repair or replacement of the gene; transduction of its product could be achieved by gene transfer, stem-cell transplantation, whole-organ transplantation in some cases, or enzyme replacement or enhancement. But for the majority of lysosomal-storage diseases that have a neurologic phenotype, none of these are yet practical.
What are the alternatives? Dr. Kolodny already has mentioned depletion of toxic substrates or metabolites, which might in theory be achieved by restriction of ingestion of substrate or its precursors; not a practical proposition for the lysosomal storage diseases as it is for the small molecule diseases. One could inhibit the synthesis, which is our topic, or in theory remove after accumulation. But again, when you are talking about macromolecules within organelles and cells, we simply don't know how to achieve that.
And finally, there is a potential for interfering or intervening in downstream pathways somewhere in the metabolic network so that one might, for example, manipulate signaling pathways to diminish apoptosis or activate alternate catabolic pathways.
Slide 15. Inhibition of Glycosphingolipid Synthesis with NB-DNJ or NB-DGJ
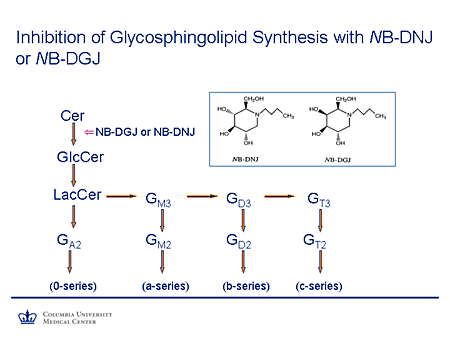
This diagram is just to remind you that the step in which a glucose molecule is added to ceramide to form glucosylceramide is the first committed step in the synthesis of all of the glycosphingolipids. This is partially competitively inhibited by NB-DNJ and its galactose congener, N-butyldeoxygalactonojirimycin (NB-DGJ), which are both essentially glucose methyl-lactose molecules with this nitrogen substituted and a small carbon side chain.
Slide 16. Miglustat: Experience in Gaucher Disease
Treatment Strategies for Liposomal Storage Diseases: Miglustat

So again, what is one aiming at? It's reduction of the stored pool of substrates shown in this cartoon. This is Gaucher disease, in which the ability to clear a substrate being synthesized at a normal rate is impaired and, thus, there is an expanded pool. By impairing the synthesis of substrate, the pool is reduced to a more manageable size.
There is evidence from studies in humans that GM1 expression is decreased by almost 40% in patients with type 1 Gaucher disease after 12 months of therapy. And again, the reduction in the substrate biosynthesis will lead to decreased accumulation and approach restoration of metabolic balance.
Slide 17. Extensive Distribution of Miglustat in Extravascular Tissues: Animal Data

Miglustat also has an advantage as a small molecule, being more widely distributed to the tissues than is the case with a large molecule such as an enzyme. Here you see the tissue-to-plasma ratios of miglustat. In liver it's 3.3 to 1. It's about 1 to 1 in the bone marrow, which is important because of the problems with bone disease, bone penetration, and enzyme replacement therapy for Gaucher disease, and there is about 30% penetration to brain.
These are data from an animal model in a rat.
Slide 18. Extensive Distribution of Miglustat in Extravascular Tissues: Human Data
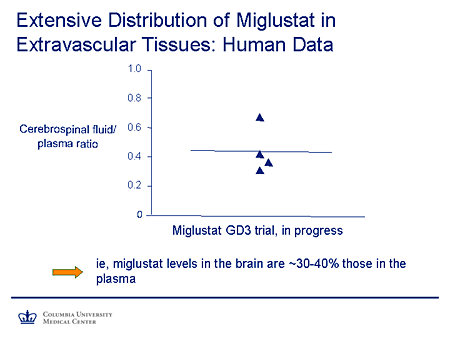
Nevertheless, we have some human data available here. This is the cerebrospinal fluid (CSF) to plasma ratio of miglustat obtained from the miglustat type 3 Gaucher disease trial. And you're seeing here a level of around 35% to 40% in the CSF compared with the plasma, which suggests a fairly significant extent of CNS penetration.
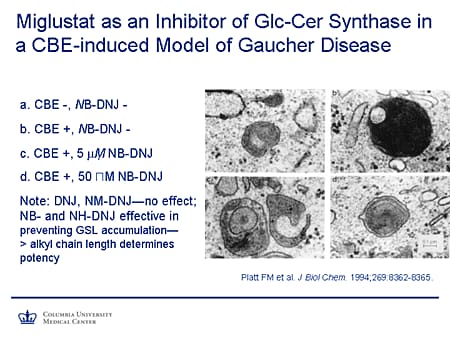
Slide 19. Miglustat as an Inhibitor of Glc-Cer Synthase in a CBE-Induced Model of Gaucher Disease
Just to give a little background, a lot of the preliminary work on the use of substrate reduction was done by Platt and colleagues in the University of Oxford, Oxford, United Kingdom, glycobiology group. They created a cellular model of Gaucher disease by treating the cells with conduritol-B-epoxide (CBE). Here in panel a, top left, you see a control cell. On the top right (panel b) is a cell that's being treated with CBE and you can see, comparing the lysosomes, the extent of storage. And this was confirmed chemically to be a glucocerebroside. After treatment with miglustat at 5 micromol and 50 micromol, there is substantial reduction in glycosphingolipid accumulation. Interestingly, and perhaps not surprisingly, there is quite a degree of specificity in terms of the alkyl chain length in these compounds that determines their potency.
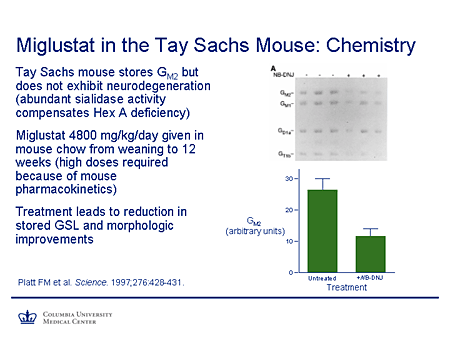
Slide 20. Miglustat in the Tay-Sachs Mouse: Chemistry
A key breakthrough was this paper from Platt's group that was published in Science in 1997. They studied the effectiveness of miglustat in the Tay-Sachs murine model. This is an interesting mouse model, which also reinforces some points about extent of substrate accumulation and clearance, because although this mouse has a pathologic phenotype and a biochemical phenotype with excess stored lipids, it does not have a clinical phenotype. So although the thresholds for those phenomena are exceeded, the threshold for production of symptoms is not. So this makes it a very nice model to study. And, in fact, the reason for this is because there is abundant sialidase activity in this mouse and that is able to partially compensate for the hexosaminidase A activity.
The mice were given very large doses of miglustat. That's required because of the pharmacokinetics of this compound in the mouse. You can see here the untreated mice. I just draw your attention to the GM2 and GM1 accumulation, which is reduced in the treated mice. The same applies for the G and T series gangliosides. Again, if one looks here at GM2 storage in arbitrary units, there is substantial reduction in comparison between the untreated and the treated mice.
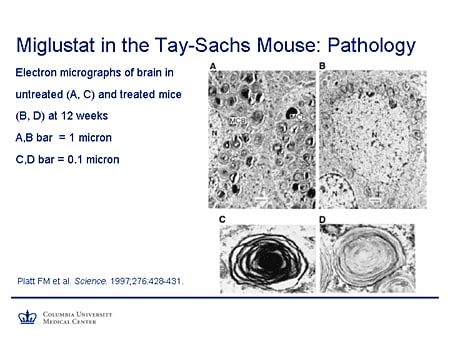
Slide 21. Miglustat in the Tay-Sachs Mouse: Pathology
The pathology also shows improvement. The panels on the left, A and C, show an untreated mouse with characteristic multilamellar cytoplasmic bodies. Now they're not entirely absent, but there is a reduction in their number and an evident reduction in the amount of substrates stored in these animals. So there is improvement both in biochemical terms and in terms of the neuropathology.
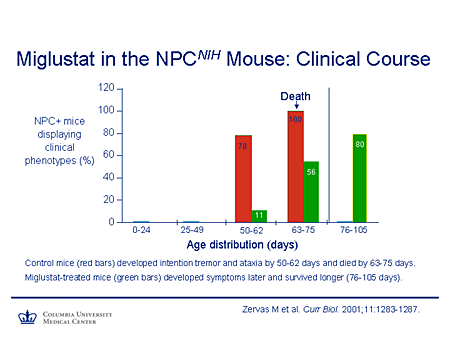
Slide 22. Miglustat in the NPCNIH Mouse: Clinical Course
This is the work of Walkley in the Niemann-Pick C mouse. This shows the number of mice (on the y-axis) that display a clinical phenotype, the x-axis age distribution. In the control mice this is a very severe null phenotype. There's 100% death within the 63- to 75-day window. The mice that have been treated with miglustat show a delay in the onset of symptoms and an increase in their survival. This is not a cure, but it ameliorates the course of disease.
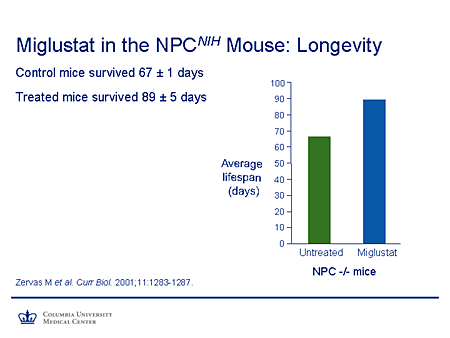
Slide 23. Miglustat in the NPCNIH Mouse: Longevity
To summarize this in a different way, you can see the change in average lifespan between the untreated and the treated mice. One might argue and say, what's 20 days or so improvement? Well if one could translate that figure from this very severe murine phenotype to humans, it could, in fact, represent a very substantial improvement in survival and also, of course, allow the opportunity for other effective therapies to be used.
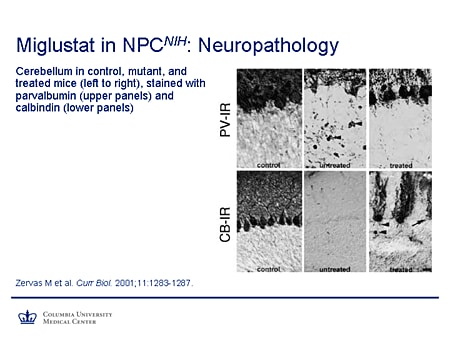
Slide 24. Miglustat in NPCNIH: Neuropathology
This shows improvement in the neuropathology in the Niemann-Pick C mouse. This is parvalbumin staining in the upper panels, calbindin staining in the lower. In the controls you can see these robust Purkinje cells filling the Purkinje cell layer. In the untreated mouse they're virtually absent in calbindin and very few left in the parvalbumin staining. And you also notice the prominent axonal spheroids, which are characteristic. In the treated mouse, there certainly is not complete rescue, but there is substantial amelioration of the neuropathologic phenotype.

Slide 25. ERT vs SRT
If we compare briefly enzyme replacement and substrate-reduction therapy, enzyme replacement is, of course, the gold standard for Gaucher disease, but it has drawbacks. It requires intravenous administration. It's dosed intermittently. There is very limited, if any, functional CNS penetration and very limited bone penetration.
Substrate reduction has the advantage of oral administration. It can be given daily. It penetrates the nervous system and appears to have better bone penetration.

Slide 26. SRT: Conclusions
So what conclusions could we draw from this? Miglustat has been approved in the European Union, the United States, Canada, and Israel for mild-to-moderate type 1 Gaucher disease when enzyme replacement therapy is unsuitable. The agent generally is well tolerated.
The major adverse effects that have been reported include weight loss, which is generally reversible; gastrointestinal side effects, particularly diarrhea, which largely can be ameliorated by dietary modification or, if necessary, the use of agents such as loperamide; and tremor, which appears to be dose related and reversible.
The animal data show promise for amelioration of neurologic disease in lysosomal storage diseases through multiple downstream effects that we've detailed.
Human studies are in progress in Niemann-Pick C (we have a poster showing preliminary results from the first year of our study in individuals 12 years of age and older with Niemann-Pick C that are quite promising), GM2 gangliosidosis, and type 3 Gaucher disease.
We emphasize these are not curative therapies, but they do have the potential to slow or stabilize neurologic disease while we await definitive treatments.
And there is certainly a likelihood that combination therapies (for example, with anti-inflammatory agents, perhaps neurosteroids in the case of Niemann-Pick disease type C) might amplify this effect, at least based on preliminary cellular and animal studies.
Epidemiology of Lysosomal Storage Diseases

Slide 1. The Role of Small Molecule Inhibition in the Balance for Inborn Errors of Metabolism
Slide 1. Small Molecule Inhibition for Gaucher Disease: Emerging Role of Substrate Reduction Therapy for Lysosomal Storage Disorders
Slide 2. Replacement Therapies for Lysosomal Diseases
As an introduction to today's program, let us consider what forms of therapy are now available or under study for the lysosomal storage diseases (LSDs). Several of these will be included in the remarks of our 3 presenters. They represent a really dramatic step forward in our quest to conquer the LSDs. They are bone-marrow transplantation; enzyme-replacement therapy; substrate-reduction therapy; stem-cell therapy; chemical-chaperone therapy, also known as enzyme-enhancement therapy; and, at least on an experimental basis, gene therapy.
Prevention is also possible, as we know, through carrier screening and family counseling and prenatal diagnosis. And the era of newborn screening certainly is close at hand.
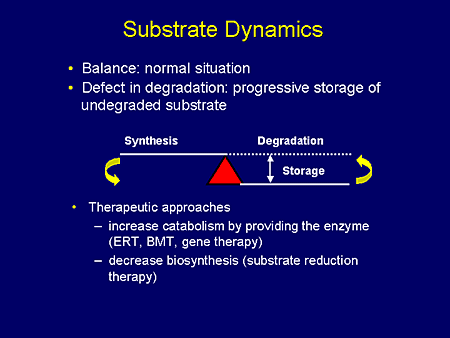
Slide 3. Substrate Dynamics
Let us now, just for a moment, focus on substrate-reduction therapy. Under normal circumstances, the amount of substrate synthesized exactly balances the amount undergoing degradation. In the LSDs, this balance is disturbed by deficiency in substrate hydrolysis, that is, in the catabolism leading to the storage of substrate.
To correct this imbalance we have at least 2 options. One is to increase catabolism by providing the enzyme through enzyme-replacement therapy, bone-marrow transplantation, or, perhaps in the future, gene therapy. A second therapeutic option is to decrease biosynthesis or, as we commonly refer to it now, substrate-reduction therapy.
Slide 4. Two Classes of SRT Molecules
Two classes of substrate synthesis inhibitors have been studied. These are the iminosugar inhibitors, of which the most well-studied is the glucose analog of N-butyldeoxynojirimycin (NB-DNJ). The second class of inhibitors is known by short terminology as the P4 or Shayman derivatives.
Most of our discussions today will focus on the glucose analog, NB-DNJ, which is known by the investigative terminology OGT 918 and has been marketed as miglustat or the tradename Zavesca. The structure of miglustat is shown here. It's a small molecule.
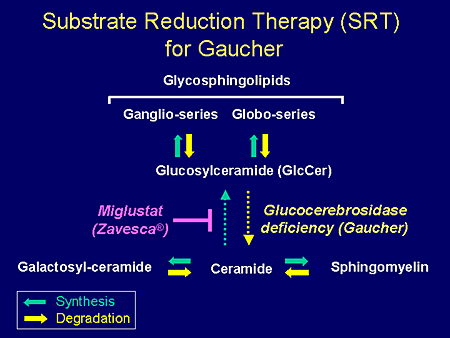
Slide 5. Substrate Reduction Therapy (SRT) for Gaucher
To just set the scene a little bit for the presenters who will follow me, bear in mind that the metabolic pathway for the synthesis of the glycosphingolipids begins with the formation of glucosylceramide from ceramide by the synthetic enzyme glucosylceramide synthase. The degradation or catabolism or hydrolysis of glucosylceramide requires the enzyme glucocerebrosidase. This enzyme is deficient in the most common of the lipid storage diseases, Gaucher disease, so that there is an accumulation of this substrate glucosylceramide. This accumulation can be blocked by the drug miglustat, which is an inhibitor of the biosynthetic pathway to glucosylceramide.







/https%3A%2F%2Fassets.over-blog.com%2Ft%2Fcedistic%2Fcamera.png)
/http%3A%2F%2Fwww.gnmhealthcare.com%2Fclients%2Fimages%2F12%2F07%2F28%2F2450011_0.gif)
/http%3A%2F%2Fanmisims.canalblog.com%2Fimages%2FLIERRE_fleur445.gif)
/https%3A%2F%2Fassets.over-blog.com%2Ft%2Fcedistic%2Fcamera.png)